分类号: Q78 编号:
本科生基因工程实验论文

指导教师: X X X
学 生: X X
专 业: 生 物 科 学
年 级: 20##
2012 年 8 月 24 日
目 录
摘要... .2
绪论... 3
材料与方法.... 4
1材料.... 4
1.1试剂及仪器.... 4
1.2菌种.... 5
1.3质粒.... 5
1.4常用溶液的配制.... 5
2方法.... 6
2.1引物的设计.... 6
2.2 NK基因的PCR扩增.... 7
2.3 DH5a和BL21(DE3)感受态的制备.... 7
2.4 pMD19-T-NK的构建及转化和鉴定.... 8
2.5 pET-trx、pET-NK、pUC-Nk的酶切与回收.... 9
2.6 pET- trx-NK的构建及转化和检测.... 11
2.7 pET-trx-NK的诱导表达.... 12
2.8 pET-trx-NK表达的SDS-PAGE检测.... 13
结果.... 15
1. NK基因的PCR扩增.... 15
2. pMD19-T-trx的构建与鉴定.... 16
3. pET-trx、pET-NK、pUC-Nk的酶切与回收.... 16
4. pET-trx-NK的构建与检测.... 18
5. pET-trx-NK的诱导表达与SDS-PAGE检测.... 19
讨论.... 20
参考文献.... 21
致谢.... 21
感想.... 22
纳豆激酶基因表达载体的构建及在大肠杆菌中的表达
摘要
纳豆激酶是一种枯草杆菌蛋白激酶。经研究发现纳豆激酶具有纤溶活性和溶栓能力, 可治疗和预防血栓病。此外,纳豆激酶还具有降低血液黏度、降血脂、降胆固醇, 改善血液循环状况, 维持血细胞的正常形态和功能等作用。本文主要介绍利用基因工程实验技术操作纳豆激酶基因,将已经连接在pMD18-T载体上的纳豆激酶酶源基因用PCR扩增出837bp的纳豆激酶的基因片段,与T载体连接后导入感受态的DH5α菌中筛选鉴定。用双酶切切下纳豆激酶的成熟基因片段,插入带有6个组氨酸标签的原核表达载体pET-trx,导入 BL(21)DE3感受态菌中诱导表达纳豆激酶蛋白质,用SDS-PAGE鉴定。
关键词:纳豆激酶,基因表达,载体,大肠杆菌
Constructing Expression Vector and Expressing nattokinase in E.coli
ABSTRACT
Nattokinase is a kind of subtilopeptidase. The study found that Nattokinase has activity of fibrinoltic which can treat and prevent thrombosis. In addition, Nattokinase can also lower blood viscosity, blood fat, cholesterol, improve blood circulation and maintain normal blood cells form and function and so on. Main of this article describe the gene engineering operation in nottokinase gene which has already connected with pMD-18-T vector amplified by PCR in which can get 837bp nattokinase gene fragments ,then linking with T vector that transform it into DH5α bacteria which were screened after identification. The mature peptide can be cutted by double-digested and insert with six histidine-tagged prokaryotic expression vector pET-trx. At last, induced the protein expression of nattokinase in the expression of E.coli BL (21) DE3. Eventually , identify that by SDS-PAGE.
KEY WORDS: nattokinase, gene expression, vector,E. coli
绪论:
纳豆激酶(nattokinase简称NK)是一种枯草杆菌蛋白激酶,是在纳豆发酵过程中由纳豆枯草杆菌(Bacillus subtilisl natto)产生的一种具有强烈纤溶活性的碱性丝氨酸蛋白酶[1]。纳豆激酶是食品纳豆中提取或纳豆菌发酵生产的分子量小的单链蛋白质。
由于纳豆激酶具有纤溶作用强,作用迅速,药效时间长;来源于食品,安全性能好,更易被人体消化吸收。可通过静脉与口服两种途径给药以及不引起出血的副作用等第一代溶栓药物所不具备的优点,使其具有开发成新一代溶栓药的潜力。
纳豆激酶于1987年由日本学者sumi在纳豆中发现。其成熟肽含有275 个氨基酸,分子量约为28ku,等电点为8.6±0.3。 从纳豆中分离和纯化出一种蛋白激酶L这种酶具有很好的溶栓作用 , 是一种枯草杆菌的发酵产物,为丝氨酸蛋白酶类,称其为 (nattokinase) L由275个氨基酸组成分子量为27kD,等电点为8.16±0.13L这种酶与其它溶栓药相比,除具有较强的溶栓作用外,还具有体内作用时间长,无毒无副作用等优点。[2]我们用 PCR 方法克隆纳豆激酶基因,并重组到表达载体上进行了初步表达及表达产物的活性测定,为用基因工程菌生产纳豆激酶奠定基础 。
经研究,纳豆激酶具有水解纤维蛋白的作用,它具有严格的限制性酶切位点,可直接作用于交联纤维蛋白,使其水解成可溶性的小分子,从而达到直接溶解血栓的作用。[3]提取纯化纳豆激酶,与交联的纤维蛋白酶解,发现不同时间降解交联纤维蛋白片段大小不同,出现1413kD、36kD、和41 kD不同大小的片段,从而白哦名纳豆激酶具有直接降解血栓的作用。纳豆激酶活体内尿激酶原转化为尿激酶,从而增加纤维酶的活性,进一步加强它的溶栓作用;纳豆激酶刺激血管内皮细胞产生内源t-PA,t-PA催化纤溶酶原转变纤溶酶,以使积累的纤维蛋白或血栓溶解;纳豆激酶通过降解好识货纤溶酶原激活剂的抑制剂(PAI-1)调控纤溶作用。[4]
纳豆中含有100种以上的酶, 碱性蛋白酶和A-淀粉酶易将纳豆中蛋白质分解便于人体消化吸收, 弹性蛋白酶( Elastase) 和植酸酶( Phytase) 目前已被从纳豆中提取生产应用于食品及其它工业中。[5]纳豆激酶具有防治心脑血管栓塞、抗致病菌、防癌、抗癌、阻止动脉粥样硬化、抗氧化、延缓衰老等多重作用,因此,纳豆激酶成为现在一项热点研究项目。
据统计, 目前美国每年有15万人死于中风,在我国,每年死于冠心病者约60万人,死于脑梗塞,脑溢血者120万人,约有80%病例是由于血栓引起而导致突发性恶果。[6]而纳豆激酶因具有特殊的溶血栓活性,既能预防也能治疗血管栓性疾病,与其他溶栓药物相比,具有安全性能好、无免疫原性、易被人体消化吸收、可静脉注射也可口服、半衰期长、生产价格低廉等优点,极有可能成为新一代李晓你哥的预防和治疗栓塞的新型药物,将为解决莫倩血栓溶解药物口服性差、价格昂贵、并发症多等问题带来更多希望。
综上所述, 我国学者对纳豆激酶基因的克隆、表达及表达产物的活性方面进行了深入系统的研究, 已经初步能够利用基因工程菌生产纳豆激酶样品。但是对于基因工程菌的发酵工艺、表达产物的分离纯化工艺、表达产物的产量及收率方面仅进行了一些探索性研究, 要想实现利用基因工程菌工业化生产纳豆激酶, 还需更加深入的研究。
材料与方法
1.材料
1.1试剂、仪器及实验用具
1.1.1仪器
台式离心机,旋涡混合器,微量移液取样器,气浴振荡摇床(不需加水),制冰机(GRANT),超净工作台(上海智城分析仪器制造有限公司),微波炉和电磁炉,电泳仪(北京市六一仪器厂,DYY-11型电泳仪)和电泳槽,电子分析天平(德国Sartorius公司),恒温水浴锅(上海博讯实业有限公司医疗设备厂出品,HSH型),紫外分光光度计,PCR仪(Biometra 华粤企业公司),通风橱,三用紫外分析仪,恒温细菌培养箱,凝胶成像仪,高压蒸汽灭菌锅(日本HIRAYAMAHVE-50高压灭菌锅)。
1.1.2实验用具
超净工作台,玻璃涂布棒,酒精灯,双面微量离心管架,试管架,记号笔,手表,摇菌试管,1.5mL离心管,0.5mL微量离心管,枪头(0~10uL,20~200uL,200~1000uL),100mL三角瓶,250mL 三角瓶,凝胶成像系统,一次性薄膜手套等,0.2mLPCR微量管,泡沫塑料保温盒,培养皿,琼脂糖凝胶电泳槽和梳子及其制胶模块, 垂直电泳槽及配套的玻璃板、夹子、密封条、梳子。
1.1.3试剂
LB 培养基, 氨苄青霉素储存液, 无菌ddwater, 100mg/mL(M/V)IPTG, 20%葡萄糖,1.0mol/L Tris HCl (pH8.8), 0.5mol/L Tris HCl( pH6.8),10%SDS,30% Acr/Bis, 10% Aps,2×上样缓冲液,TEMED ,低分子量蛋白分子量 Marker,考马斯亮蓝染色液, 5 X电泳缓冲液,20%(M/V)IPTG,2%(M/V)X-gal,50 X TAE电泳缓冲液,溴化乙锭储存液,10×加样缓冲液,6×加样缓冲液,DNA分子量Marker,电泳级琼脂糖粉,3U/μL Taq DNA 聚合酶,10×PCR 缓冲液(MgCl2 free),25mM MgCl2,dNTP 混合液, 10mg/mL RNase A,TE缓冲液,95%和 70%乙醇,EcoR I 和 BamH I 限制性内切酶,10×内切酶缓冲液,0.1 mol/L CaCl2溶液,T4 DNA连接酶,连接酶缓冲液。
1.2菌种
带有 pET-trx 质粒载体DH5α菌,带有pMD18-T-NK载体的DH5α菌, E.coli DH5α,E.coli BL21(DE3)。由基因工程实验室提供。
1.3 质粒
1.3.1目的基因所在质粒:pMD-18T-NK

1.3.2表达载体:pET-trx

1.4 常用溶液的配制
1.4.1 氨苄青霉素储存液:无菌水配制 100mg/mL,分装后-20?C保存。
1.4.2 LB 培养基:胰化蛋白胨 10g,酵母提取物 5g,NaCl 10g,加 200mL ddwater搅拌完全溶解,用约NaOH 调 pH 至 7.0,加 dd water 至 1L,分装4个200ml和2个100ml,121℃ 20min 灭菌。使用时可加入氨苄青霉素,终浓度100ug/mL。
1.4.3 溶液Ⅱ(NaOH/SDS溶液):0.2mol/L NaOH,1%SDS,现用现配。
1.4.4 50×TAE电泳缓冲液(pH约8.5):Tris碱242g,57.1ml冰乙酸,37.2g Na2EDTA·2H2O,dd water 定容至1L。使用时稀释成1×。
1.4.5 琼脂糖凝胶:称取0.25g琼脂糖粉,放入三角瓶,加入25ml 1×TAE 电泳缓冲液(注意原液是50×,按照1:49的比例稀释),用保鲜膜盖住,放入微波炉里烧开(30 s),取出观察,若无颗粒则置于桌面上冷却至不烫手即可灌胶,若仍然有颗 粒状物,需要再继续加热,直至无颗粒后冷却灌胶。
1.4.6 1.0mol/L Tris HCl pH8.8:称取12.1g Tris碱, 加50mL蒸馏水,缓慢地加浓盐酸至pH8.8(约加 8mL)。让溶液冷却至室温,pH将会升高,然后再调至 pH8.8,加蒸馏水至 100mL。高温高压灭菌后 4℃保存。
1.4.7 0.5mol/L Tris HCl pH6.8:称取 6.05g Tris 碱溶于 40mL 蒸馏水中,加约 48mL 1mol/L HCl,让溶液冷却至室温,再用 HCl 调至 pH6.8,加水稀释到 100mL 终体积。高温高压灭菌后 4℃保存。
1.4.8 30% Acr(丙烯酰胺)/Bis(N,N’-亚甲基双丙烯酰胺):30g Acr+0.8g Bis,用 dd water定容至100mL,4?C棕色瓶避光保存(现用现配)。
1.4.9 10% Aps(过硫酸铵):蒸馏水配制,-20℃保存。过硫酸铵会缓慢分解,一周内使用完。
1.4.10 上样缓冲液:0.5mol/L Tris HCl pH6.8 2mL,甘油 2mL,20% SDS 2mL,0.1%溴酚蓝0.5mL,β-2-巯基乙醇1.0mL,dd water 2.5mL,室温存放。
1.4.11 电泳缓冲液:Tris 7.5g ,甘氨酸36g,SDS2.5g,加ddwater 至500mL。使用时稀释5倍。
1.4.12 考马斯亮蓝染色液:考马斯亮蓝 R250 0.25g + 45mL 甲醇+10mL 冰醋酸,用ddwater 定容至100mL 。
1.4.13 LB 平板培养基:LB 培养基中含 1.2 %(1.2g/100ml)琼脂,高压灭菌后加入氨苄青霉素至100μg/mL,每个培养皿中约 20mL 倒制平板。
2. 方法
2.1 设计引物
PCR引物:
上游引物(PDU ,5’端带BamHI酶切位点):
5'-GGATCC ATG GCC TCC TCC GAG AAC-3'
下游引物(PDL,5’端带EcoRI酶切位点):
5'-GAATTC TAC AGG AAC AGG TGG TG -3'
2.2 NK基因的PCR扩增
2.2.1 由老师提供pMD-18-T-NK质粒进行PCR扩增。
2.2.2 在 0.2mL PCR微量离心管中按下列剂量配制 50μL 的PCR反应体系。

2.2.3 设置 PCR 仪的循环程序:
 ① 94℃ 5min
① 94℃ 5min
 ② 94℃ 30s
② 94℃ 30s
③ 60℃ 30s
④ 72℃ 1min
 ⑤ 72℃ 10min
⑤ 72℃ 10min
PCR总时间需要 1h50min。
PCR结束后,取5μl产物进行琼脂糖凝胶泳。观察是否有预计分子量的主要产物带。
注:退火温度
NK:60℃ 红色荧光蛋白:51℃ 绿色荧光蛋白:61℃
2.3 DH5α和BL21(DE3)感受态的制备
2.3.1 取2个1.5mL 预冷10 min的无菌微量离心管,做上相应标记。
2.3.2 其中一管加入1.0mL BL21(DE3)菌液,另外一管加1.0mL DH5α菌液,然后在冰上放置 10min, 5000r/min 4℃ 离心 10min,弃上清。
2.3.3 加入 1ml 冰冷的 0.1 mol/L CaCl2溶液,立即在涡旋混合器上混匀,插入冰中静置 30min。
2.3.3 4℃,5000r/min 离心 10min。弃上清液后,用 1mL 冰冷的 0.1 mol/L CaCl2溶液重悬菌体,插入冰中放置 30min。
2.3.4 4℃,5000r/min 离心 10min。弃上清液后,用 200uL 冰冷的 0.1 mol/L CaCl2溶液重悬菌体。
2.3.5 在超净工作台内将两种菌按50ul/管分装在1.5ml管内,放于4℃冰箱待用。
2.4 pMD19-T-NK的构建及转化和检测
2.4.1 PCR 产物(NK)与pMD-19-T (PCR 2.1)载体直接连接
阳性PCR产物与pMD-19-T载体(PCR 2.1)连接。在离心管中加入:

在干式恒温气浴中14℃,连接3 h。
2.4.2 感受态细菌的转化
2.4.2.1 将恒温水浴的温度调节到 42℃。
2.4.2.2 从4℃ 冰箱中取出感受态菌,冰浴7min。
2.4.2.3 取5 uL连接好的质粒与感受态细菌DH5a混匀,在冰上放置20min。
2.4.2.4 于42℃水浴中90S进行热休克,然后在冰中放置4 min,在超净台上加入300uL LB培养基(不含氨苄青霉素),37℃振荡培养45 min。
2.4.2.5 超净台上取上述转化的混合液150μL、200μL于新的1.5ml的离心管中,加入40μL 20% x-gal,7μL 20% IPTG 混匀。
2.4.2.6 将上述菌液用涂布棒均匀的涂布在Amp+LB 的固体培养基上,37℃恒温培养过夜。
2.4.3 pMD-19-T-NK的鉴定(提取质粒使用Axygen试剂盒)
2.4.3.1 观察菌落生长状态,用10uL枪头挑取3个白菌落分别置于含1.7mL LB培养基中,37℃摇菌培养8h。
2.4.3.2 分别取 1.4mL 的菌液于 1.5mL 离心管中,10000r/min 离心1min,弃上清。
2.4.3.3 加250ul Buffer S1悬浮细胞沉淀。
2.4.3.4 加250ul Buffer S2温和上下翻转6-8次,12000r/min离心10min。
2.4.3.5 吸取上步中离心上清液并转移到制备管,12000r/min离心1min,弃滤液。
2.4.3.6 将制备管置回离心管,加500ul Buffer W1,12000r/min离心1min,弃滤液。
2.4.3.7 将制备管置回离心管,加700ul Buffer W2,12000r/min离心1min,弃滤液。
2.4.3.8 方法同上制备离心管,加700ul Buffer W2,12000r/min离心1min,弃滤液。
2.4.3.9 将制备管置回2ml离心管, 12000r/min离心1min。
2.4.3.10 将制备管移入新的1.5ml离心管中,在制备管中加60-80ul Eluent,加热至65℃后,室温静置1min, 12000r/min离心1min。
2.4.3.11 电泳检测:以蓝菌落作对照,电泳检测T4-NK是否连接并转化成功。
2.5 pET-trx、pET-NK、pUC-Nk的酶切与回收
2.5.1 提取pET-trx、pET-NK 及pUC -NK的质粒(小提质粒Axygen试剂盒)
2.5.1.1 取菌液10000r/min 1mim离心,弃上清。
2.5.1.2 向吸附柱cp3中加入500μL的平衡液BL,12000r/min 1 min,弃废液。
2.5.1.3 将菌体沉淀的离心管中加入250μL的溶液P1进行悬浮。
2.5.1.4 离心观众加入250μL的P2,将离心管温和上下翻转6-8次,混匀,使菌液达到清亮粘稠。(此步不超过5min)。
2.5.1.5 离心管中加入350μL的P3,温和翻转6-8次,混匀,12000r/min 10min。
2.5.1.6 将上清转移到吸附柱cp3中,12000r/min 60s ,弃废液。
2.5.1.7 cp3中加入600μL漂洗液pw(无水乙醇),12000r/min 60s 。弃废液。
2.5.1.8 重复上步。后12000r/min 2min离心 。弃废液。
2.5.1.9 将吸附柱在新的离心管中,向膜中心滴加100μL洗脱buffer EB,放置2min后,12000r/min 2min离心。
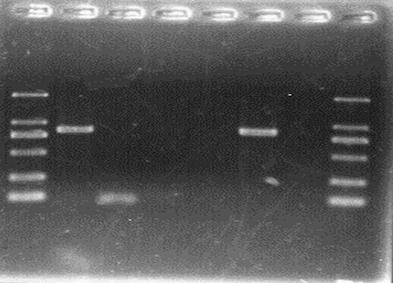
2.5.1.10 琼脂糖凝胶电泳检测提取质粒的质量。点样顺序1-7孔分别为张婷组酶切产物、pET-NK(广)、pUC-NK(广)、pET-trx(广)、pET-NK(田)、pUC-NK(田)、 pET-trx(田)。
2.5.2 pET-trx、pET-NK、pUC-NK 双酶切
2.5.2.1 pET-trx质粒 DNA双酶切体系:

2.5.2.2 pET-NK质粒 DNA双酶切体系:

2.5.2.2 pUC-NK质粒 DNA双酶切体系:

2.5.3 pET-trx、pET-NK、pUC-NK胶回收
2.5.3.1 用 1×TAE 和琼脂糖配制 1%回收胶。
2.5.3.2 从恒温水浴锅中取出酶切产物,分别加入4uL的10× Loading Buffer终止反应,并混匀。
2.5.3.3 选择13个相邻的点样孔,按顺序分别加入pET-trx空质粒,pET-trx大片段(小样),pET-trx大片段(大样-广),pET-trx大片段(大样-田),菌落质粒,PCR产物,pET-NK(小样)、pUC-NK(小样),空泳道,pET-NK(大样-广),pET-NK(大样-田),pUC-NK(大样-广),pUC-NK(大样-田)
2.5.3.4 电泳结束后用电话卡将含有大量酶切产物的泳道切下,EB 染色含小样的胶块。在紫外灯下找到目的 DNA 带,用刀片在目的 DNA带的下上下边缘各切一个小口作为标记。
2.5.3.5 将做好切口标记的凝胶与未染色的凝胶原位对齐,根据小胶条上的切口标记估计未染色的胶块上大样的 DNA 酶切产物的位置。
2.5.3.6 用刀片切下大胶中 DNA 产物所在的凝胶(尽量不要多余的凝胶),分别转移至一个事先称量过的、灭活的 1.5mL 微量离心管中。再称量后计算出其中胶块的重量。
2.5.3.7 再用 EB 染色切除 DNA 以后的大胶,与小胶条并在一起在紫外灯下检查是否已把 DNA 带切走。
2.5.3.8 按照 DNA 凝胶回收试剂盒的说明书操作,回收 DNA 酶切产物。
2.5.3.8.1 向吸附柱CA2中加入500μL 的BL,12000r/min 1min 离心,弃废液,将吸附柱从新回收到收集管中。
2.5.3.8.2 将单一的目的DNA条带胶块的离心管中加入600μL的溶胶剂。
2.5.3.8.3 在60℃水浴锅中放置10 min,中间隔2-3 min轻晃几次,确保胶体完全融化,没有胶体颗粒剩余。
2.5.3.8.4 在溶胶过程中,平衡柱子,取6个有膜的ep管,加入500μL的平衡液BL,12000r/min 1min 离心。
2.5.3.8.5 待溶胶好后,静止2min(由于温度过高,平衡柱结合DNA能力较弱),12000r/min,离心60s。
2.5.3.8.6 上柱,离心后,弃废液。想cp柱中加入600μL,漂洗液pw,12000r/min 60s。弃废液。
2.5.3.8.7 重复上步。弃废液,12000r/min,2min,弃废液。开盖放置,彻底晾干。
2.5.3.8.8 将CA2,加入新的ep管中,先加入洗脱缓冲液EB 350μL,放置2min。12000r/min 2min。收集DNA洗脱液。
2.5.3.9 pET-trx NK gene 胶回收电泳验证
2.6 pET-trx-NK的构建及转化和检测
2.6.1 pET- trx -NK的构建
(1) 提前将干式恒温仪(或泡沫塑料保温盒里的水)温度调整到 14℃。
(2) 取一个高压灭活的 0.5mL 微量离心管,加入如下连接体系:

将上述混合液轻轻震荡后再短暂离心,然后置于 16℃水中保温过夜连接。
2.6.2 pET-trx-NK转化BL21(DE3)
2.6.2.1 将恒温水浴的温度调到 42℃。
2.6.2.2 从4℃冰箱中取出一管BL21(DE3)感受态菌,插入冰上。
2.6.2.3 加入5μL 连接好的pET-trx-NK质粒混合液,轻轻震荡后放置冰上 25min。
2.6.2.4 轻轻摇匀后插入 42℃水浴中90s进行热休克,然后迅速放回冰中,静置 5 min。
2.6.2.5 在超净工作台中向上述管中加入300uL LB(不含氨苄青霉素) 培养基轻轻混匀,然后固定到摇床的弹簧架上 37℃震荡 45min。
2.6.2.6 在超净工作台中取上述转化混合液300ul,混匀后滴到含合适抗菌素(Amp+)的固体 LB 平板培养皿中。从酒精中取出玻璃涂布棒,在火上点燃,熄灭后稍等片刻,待其冷却后轻轻涂布均匀。
选两组不同条件进行对照

2.6.2.7 在涂好的培养皿上做上标记,先放置在 37℃恒温培养箱中约30min 直到表面的液体都渗透到培养基里后,再倒置过来放入 37℃恒温培养箱中培养过夜。
2.7 pET-trx-NK的诱导表达
2.7.1 观察平板上菌落生长状态,发现少数,不明显的菌落散布,由于菌落的数量过少,故用其他小组的菌落进行扩培。
2.7.2 将LB培养基分装到100ml的三角瓶中,后加入氨苄青霉素(100mL/100μL)。
2.7.3 将加好(Amp+)的LB培养基分装到灭菌的试管中1.6mL/管
2.7.4 挑菌,37.4℃摇床中,进行摇菌扩培。
2.7.5 质粒提取使用Axygen试剂盒,步骤与2.5.1小提质粒相同。
2.7.6 提质粒后进行电泳检测。
2.7.7 将LB培养基配制成含0.2%的LB-葡萄糖培养基(Amp+浓度为120μg/ml,即100ml/120μl的比例加入Amp+)
2.7.8 将正确的质粒对应的菌液加入6ml的LB-葡萄糖培养基培养基30℃,慢摇过夜。
2.8 SDS-PAGE检测pET-trx-NK的表达
2.8.1 电泳槽的组装及配胶
2.8.1.1 组装电泳玻璃并用细橡胶管密封底部和侧面,然后用夹子夹住玻璃两侧的中央部位。插入梳子后在玻璃上距离梳子齿底部 1cm处做一个标记。拔掉梳子,倒入自来水检验是否漏水。如不漏水,弃掉自来水后在卫生纸上倒置片刻,尽可能流尽水份。
2.8.1.2 配制 10%分离胶:按顺序和量取下列溶液混在一个 50mL 的小烧杯中:
Acrylamide-Bis 3.96ml
1M Tris(pH8.8) 4.38ml
dd water 3.432ml
10% SDS 118.8μL
TEMED 9.9μL
10%APS 118.8μL
2.8.1.3 加完TEMED 后拿起烧杯轻轻转动几下底部,将溶液混匀后,立刻缓缓倒入玻璃夹缝中,直到液面与所作的标记齐平。剩下的胶液留在小烧杯中,倾斜放置。
2.8.1.4在胶液面上部用 1000uL 微量移液器轻轻地沿玻璃壁来回移动加满 dd water。静置约 30min。直到烧杯中剩余的胶液凝固。倒出蒸馏水。
2.8.1.5 配支持胶:按顺序和量取下列溶液混在一个 50mL 的小烧杯中:
Acrylamide-Bis 0.675ml
0.5M Tris(pH6.8) 0.563ml
dd water 3.165ml
10% SDS 45μL
TEMED 7.5μL
10%APS 45μL
2.8.1.6 加完TEMED 后拿起烧杯轻轻转动几下底部,将溶液混匀后,立刻倒入玻璃夹缝中,液面与玻璃上边缘齐平。然后再慢慢插入梳子,静置约 20min。烧杯中剩下的胶液倾斜放置,直到其中的溶液凝固。
2.8.1.7去掉橡胶管,将装有胶的玻璃和梳子翻转,贴在电泳槽侧面,用夹子把玻璃两边的中部与电泳槽中部夹在一起。
2.8.1.8 在上面的槽中加满自来水试验一下是否漏水,然后倒掉自来水。
2.8.1.9 配制1X加样缓冲液400mL,从上面的电泳槽倒入,若不从下面的电泳槽漏水,则加满缓冲液,在将下面的电泳槽也加满电泳缓冲液,小心缓慢地拔出梳子。
2.8.2 外源基因的诱导表达
2.8.2.1 超净台中接种 ① pET-trx-NK的BL21(DE3)菌
② 空pET-trx转化BL21(DE3)菌
③ BL21(DE3)空菌。 37℃摇菌。
2.8.2.2 IPTG(测OD600 =0.4 – 0.6 即能诱导表达)

2.8.2.3 150-170r/min 摇菌,设置浓度梯度和时间梯度对比。
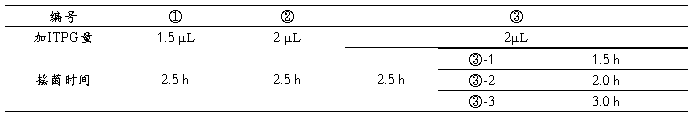
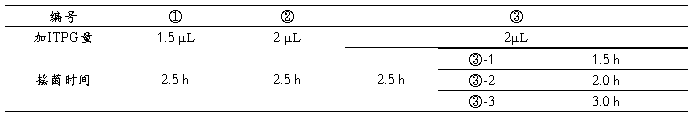
注:其中①、②、③ 菌形成等时间的浓度梯度对照,③-1、③-2、③、③-3菌形成等浓度的时间梯度对照。
2.8.3 各取1.5ml菌液及未加IPTG诱导的菌液4000rpm离心15min,弃上清,收获菌体。将表达后的菌体与2×上样缓冲液按1:1混匀于微量离心管中,用20uldd water重悬菌体,加入20ul的2×加样缓冲液,煮沸5min。
2.8.4 电泳泳道设计1-10泳道加样顺序依次为:marker、无IPTG空菌BL21(DE3)、IPTG空菌BL21(DE3)、①、②、③、③-1、③-2、③-3、无IPTG pET-trx-NK 。
2.8.5 电泳时,10 mA恒流,120V电压电泳。直至到条带到支持胶处,改为20 mA
点流,120V电压电泳。
2.8.6 染色考马斯亮蓝染色30min,流水冲掉染料后脱色液脱色1h,去离子水浸泡过夜。
实验结果与分析
1. NK基因的PCR扩增
 将实验室提供的pMD-18-T-NK质粒进行PCR扩增,在30个PCR循环中,得到NK基因的扩增片段,进行琼脂糖凝胶电泳得到图1的电泳图。
将实验室提供的pMD-18-T-NK质粒进行PCR扩增,在30个PCR循环中,得到NK基因的扩增片段,进行琼脂糖凝胶电泳得到图1的电泳图。

1.1 点样孔2-5中,点样的顺序依次为刘杭州组的NK PCR Product、我组的NK PCR Product、我组的NK PCR Product 阴性对照,田文嘉组的NK PCR Product、田文嘉组NK PCR Product阴性对照。
1.2 分析:其中我组没有NK PCR Product。
1.2.1 在加PCR体系的过程中,由于许多小组一起混用Primer1和Primer2,不能避免有污染的可能性。
1.2.2 PCR体系加样过程中,在离心管上所做的标记已经模糊,不能排除体系加混乱的可能性。
2. pMD19-T-NK的构建及转化和检测
2.1 用已经扩增好的PCR 产物(NK)与pMD-19-T (PCR 2.1)载体直接连接。并将连接好的pMD19-T-NK质粒导入DH5α感受态菌中,进行诱导鉴定。将转化的DH5α感受态菌,摇菌培养后涂平板。涂到LB(Amp+)的固体培养基中,过夜培养,进行蓝白斑鉴定。
2.2 分析:整个大组都没有培养出菌落
2.2.1 pMD-19-T载体是有实验室提供,而且全部试验小组菌未有蓝白菌落的出现。所以不能排除,pMD-19-T载体质粒出现了问题。
2.2.2 在加入LB培养基,进行摇菌培养时,LB培养基出现问题,即在摇菌时使用的是LB(Amp+)的培养基。由于pMD-19-T-NK刚导入DH5α感受态菌中,导入量很小,能够抗Amp+的菌中很少,所以用LB(Amp+)的培养基,摇菌会使大量的菌体死亡而导致涂平板时,没有菌落生成。
3. pET-trx、pET-NK、pUC-Nk的酶切与回收
3.1 pET-trx、pET-NK、pUC-Nk质粒提取电泳鉴定

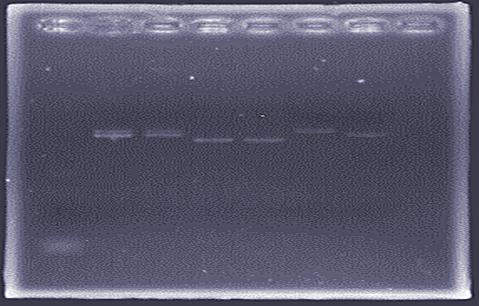 上一步中,整体大组没有转化得到所需要细菌,无法直接提取质粒,老师提供之前实验室保存的菌种pET-trx 、pET-NK、pUC-NK进行质粒的提取,然后电泳鉴定。
上一步中,整体大组没有转化得到所需要细菌,无法直接提取质粒,老师提供之前实验室保存的菌种pET-trx 、pET-NK、pUC-NK进行质粒的提取,然后电泳鉴定。
根据pUC-NK、pET-trx、pET-NK的DNA链的长度分别为3.6Kb、3.3Kb、4.1Kb,进行组内对比,分析三种质粒电泳条带所处的位置,可得出三种质粒提取物,是正确的。
3.2 pET-trx、pET-NK、pUC-NK胶回收
对所提取的三种质粒进行双酶切,过夜后进行电泳胶回收,质粒DNA。


分析:
可以从电泳图中看到1、2泳道中的pET-trx质粒和pET-trx大片段小样的电泳条带,可以说明质粒提取成功,以及酶切成功;在5-8泳道中隐约可见菌落质粒、PCR产物和两种小样的电泳条带,根据电泳条带,对应可将10-13泳道中回收的DNA条带切下回收。电泳条带的亮度不够,说明点样的浓度偏低,说以切胶的时候要尽量将小样对应的大样切下回收,减少DNA片段的损失。
3.3 pET-trx NK gene 胶回收电泳验证
利用DNA回收试剂盒,将胶体中个DNA片段回收,检测是否回收到了所需要的DNA片段。以下表中的点样顺序进行电泳。可得到图4的电泳图。

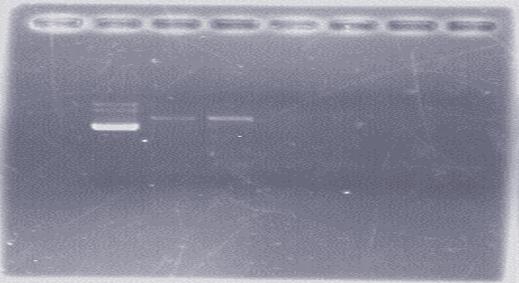
分析:
从图中可看出1泳道三条带,应为质粒较长时间保存后形成的三种构型(有上到下分别为:超螺旋、环状、线性质粒),且2泳道中的pET-trx与4、5泳道中的pET-NK 的位置一致,说明胶回收成功。
 4. pET-trx-NK的构建及转化和检测
4. pET-trx-NK的构建及转化和检测
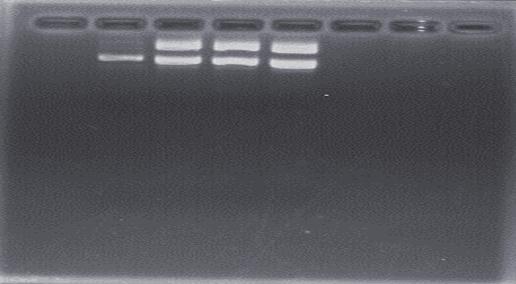
分析:
图5反应了pET-trx-NK转化菌的电泳条带。但是电泳条带并不完美,所有条带都处于点样孔的附近,并没有随着电流移动很大的距离,没有办法很明确的判断出是否确实是将pET-trx与NK基因连接。产生这样条带的原因可能是由于在配置电泳胶的时候,琼脂的质量过大,而导致电泳胶浓度偏高。或者也可能是,在配置电泳胶时候,没有将50xTAE稀释,直接用于电泳胶的配置而导致,出现图5中条带与考马斯亮蓝分离过大,即DNA条带在点样孔附近。
5. pET-trx-NK构建及SDS-PAGE检测pET-trx-NK的表达
观察平板上菌落生长状态,发现少数,不明显的菌落散布,由于菌落的数量过少,故用其他小组的菌落进行扩培。将正确的质粒对应的菌液加入6ml的LB-葡萄糖培养基培养基30℃,慢摇过夜后测定OD600 值达到一定程度后可以进行SDS-PAGE电泳检测,检测如图6所示。
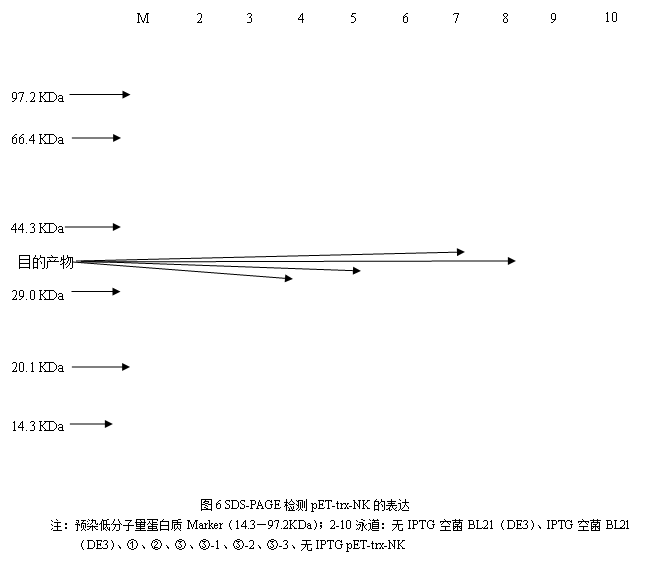
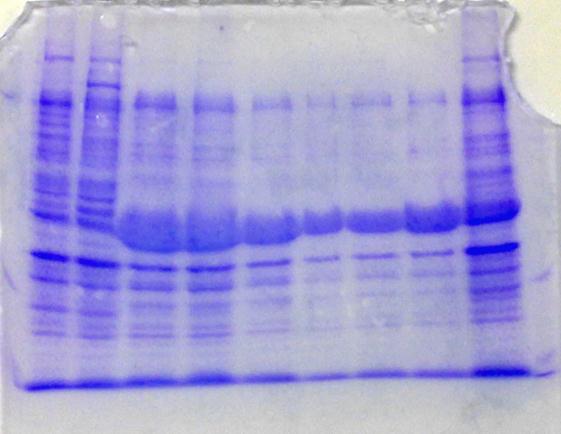

分析:
电泳点样时总共点样10,电泳完成后,只有九个条带。电泳样图呈笑脸状,Marker在第一泳道,电泳时偏离泳道而造成Maker条带的消失。但是根据标准的预染低分子量蛋白质Marker(14.3—97.2KDa)标注对照下,在Marker 44.3KDa附近以下存在着蛋白条带为目的蛋白条带,说明诱导成功。
在其他泳道中,2至10泳道:无IPTG空菌BL21(DE3)、IPTG空菌BL21(DE3)、①、②、③、③-1、③-2、③-3、无IPTG pET-trx-NK。做了时间梯度以及浓度梯度的对照,其中①、②、③ 菌形成等时间的浓度梯度对照,③-1、③-2、③、③-3菌形成等浓度的时间梯度对照。
从图中可以看出,在3至5泳道中,4泳道的目的蛋白条带相对于其他两个颜色较深,3泳道的目的蛋白条带比5泳道的颜色深,体现了②>①>③的浓度梯度对照。
在5至8泳道中,8泳道的目的蛋白条带相对于其他连个较深,然后5、7、6泳道的染色依次变浅,体现了③-3>③>③-2>③-1的时间梯度的对照。
讨论:
本次实验过程进行的比较顺利,所得到的电泳图也较好,能够清楚的鉴别是否是所需要的PCR产物以及其他的产物。
在第一次进行PCR体系添加时,由于多种原因而导致没有PCR产物(NK片段)。在后来的实验中,pMD19-T-NK的构建时,可能由于pMD19-T质粒不是很好,或者在摇菌时使用了添加氨苄青霉素的LB培养基而导致这个大组的pMD19-T-NK的构建菌没有长出菌落,而没有提取到pMD19-T-NK的质粒。
之后的实验过程中比较顺利,但是在构建pET-trx-NK感受态菌过程中,由于时间较短的原因,产生出的菌落很少且很小,故借用其他小组的菌落进行接下来的实验。最终pET-trx–NK转化表达细胞,诱导表达后,蛋白质点用检测较为成功,得到了目的基因表达的蛋白产物。
综上分析的原因,我想在以后的试验中至少能总结出几条经验教训:提高自己的实验技巧和基本功,在点样、溶胶、加PCR体系、配置各种溶液等各种基本操作中不会出错。在实验过程中,需要小心谨慎,要对每一步做好记录,对所需要做标记的离心管、ep管等随手做好标记,否则弄混以后,实验将功亏一篑。如果实验出现问题及时补救,如果不能补救则重来,切不可存在侥幸心理,否则只会浪费时间和精力。
参考文献
[1] 江晓,董明盛. 纳豆、纳豆激酶与人体保健[J].专论与综述.
[2] 刁建中,陈桂光,马波.纳豆激酶的最新研究进展[J].轻工科技. 2012.3 ,3:7-8
[3] 肖美燕,徐尔尼.纳豆激酶的提取研究[J]. 食品工业技.2007.11.28,(1):197-199.
[4] 余榕捷,汪炬,谢秋玲,洪岸.纳豆激酶酶原基因和纳豆激酶基因的克隆及在大肠杆菌中表达[J]. 生物技术.2002,12(3):2-4
[5] 王 伟,甘露,甘旭华,陈晓琳,倪敬田,唐欣昀.纳豆激酶基因的克隆和高效表达[J].安徽农业大学学报,2012,39(4):1
[6] 李莹,陈斌,敬俊锋,何正波.纳豆激酶基因的克隆及其在大肠杆菌中的表达[J]. 重庆师范大学学报(自然科学版).2012,29(1):78-82
[7] 邢万金,基因工程实验指导,内蒙古大学生命科学学院生物基因工程研究室,2007.05
[8] 张立全,刘慧,苏慧敏等.枯草杆菌纳豆激酶基因的克隆及其在E.coli BL 21(DE3) plysS中的表达[J]. 内蒙古大学学报(自然科学版),2005,5(36):284-287
致谢
感谢XX老师对我们实验的教学与指导,让我们通过基因工程的实验巩固了基因工程课堂教授的理论知识。感谢老师耐心讲解与不厌其烦的教给我们各种实验所需仪器设备的使用方法,陪我们大家一起早起晚归,其认真的态度也感染了我们大家,老师告诉我,不论实验成功与失败,我们都应该注重这个学习过程,只有不断的学习才能让你真真掌握其中的精髓。
其次感谢XX同学为大家LB配培养基,XXX、XXX同学为大家制备感受态细胞做准备工作,XXX同学配置的CaCl2溶液,有了他们的无私付出,成就了我们大组的实验顺利完成。
再次感谢在实验过程中为我小组提供NK PCR产物的XXX和XXXXX同学以及pET-trx-NK构建过程中给我小组提供菌落的XXX同学,同时感谢同组的XX同学,在实验过程中的相互合作、相互体谅,有了他们的无私帮助,才能使我小组的实验顺利完成。
感想
本次实验虽然有成功也有失败,但是在实验过程中我们学到了很多。实验让我们体会到了理论与实践相结合是一件多么不容易的事,要想保证做出理想的理论化的结果,即使在书本中一句简简单单的证实,都必须认真做好每一步,付出加倍的努力,还要怀着一颗积极的心态去完成每一个环节,不能想当然得去做实验。此外,实验中的一些细节一定不能忽略,我们最终的重大错误,往往是因为一个小小的细节而酿成的。
总结实验整个过程,我体会到,要想做好一件事,必须全身心的投入其中,认真应对每一个环节。做实验前要做好预习,明白每一个步骤以及每一步的作用,宏观的把握,这样才能保证在实验时不盲目,有一个清晰的思路;实验过程中一定要注意细节,做实验就如同做科学研究,是一个十分严谨的工作,我们必须打起十二分的精神,努力做到百分之百的正确率;合作精神。小组成员只有相互协作,相互提醒才能确保实验的正确性和高效性。