
实验报告
实验项目名称:基因工程综合实验
实验项目性质:综合性实验
所属课程名称:基因工程
班 级:11生物工程1班
学 号:
指 导 老 师 :
实验完成时间:
目录:
1.摘要 ..................................................3
1.前言 ..................................................3
2.实验材料和仪器................................................3
2.1 实验材料
2.2实验仪器 .
3.实验试剂 ..........................................4
3.1 DNA提取所需试剂
3.2 PCR实验所需试剂
3.3 双酶切实验所需试剂
4.实验步骤 ...........................................4
4.1 质粒DNA提取
4.2 聚合酶链式反应(PCR)
4.3 质粒DNA的双酶切分析
4.4 琼脂糖凝胶制备
5.实验结果与分析 ...........................................7
5.1质粒DNA提取所得凝胶电泳结果
5.2 PCR扩增实验结果
5.3质粒DNA的双酶切分析结果
摘要:实验包括质粒DNA的提取、DNA的凝胶电泳、质粒DNA中靶基因的酶切分析及质粒DNA中重组进的靶DNA序列的PCR扩增。通过本综合实验,帮助大家理解质粒DNA的提取原理、凝胶电泳中DNA分离的机理、限制性内切酶的工作原理及PCR是如何实现DNA扩增的,通过实验同时让同学掌握DNA的提取技术、凝胶电泳技术、DNA酶切分析技术及靶基因的体外快速扩增技术,了解实验中出现的现象并学会分析与解决实验中出现的有关问题。
聚合酶链反应(PCR)是体外酶促合成特异DNA片段的一种方法,由高温变性、低温退火(复性)及适温延伸等几步反应组成一个周期,循环进行,使目的DNA得以迅速扩增,具有特异性强、灵敏度高、操作简便、省时等特点。它不仅可用于基因分离、克隆和核酸序列分析等基础研究,还可用于疾病的诊断或任何有DNA,RNA的地方.聚合酶链反应(Polymerase Chain Reaction,简称PCR)又称无细胞分子克隆或特异性DNA序列体外引物定向酶促扩增技术。
关键词:凝胶电泳 限制线内切酶 DNA质粒
1 前言 本次实验对象为含有重组了1kb DNA片段的PET32.a表达质粒。该表达质粒中长度约6kb。首先采用离心破碎法破碎细胞,再使用碱裂解法提取质粒DNA。最后通过各种化学抽提纯化质粒DNA,最后得到纯化后的DNA质粒作为之后实验的材料。在碱性溶液中,双链DNA氢键断裂,DNA双螺旋结构遭破坏而发生变性,但由于质粒DNA分子量相对较小,且呈环状超螺旋结构,即使在高碱性pH条件下,两条互补链也不会充分分离,当加入中和缓冲液时,变性质粒DNA又恢复到原来的够型;而线性的大分子量细菌染色体DNA则不能复性,与细胞碎片、蛋白质、SDS等形成不溶物,通过离心沉淀可被除去,而质粒DNA及小分子量的RNA则留在上清液中。混杂的RNA可用RNaseA酶消除,再用酚/氯仿处理,可除去残留的蛋白质,达到纯化质粒DNA的目的。
PCR(聚合酶链式反应)是根据DNA双螺旋结构在变性温度下解链为单链DNA,在退火温度下加入反应体系的特异引物根据碱基互补配对原则与单链DNA特异结合,然后在延伸温度下,通过DNA聚合酶的聚合作用,不同的脱氧核苷酸按照碱基互补配对原则,由引物引导合成出与模板DNA互补的新链,实现DNA的扩增的技术。 本次实验我们将以纯化后的质粒DNA作实验材料,在预先设计好引物后用PCR技术扩增目的片段。
在PCR实验的同时,我们将利用提取的质粒DNA作目的序列的双酶切实验。限制性内切酶能在适当温度、pH等条件下识别DNA序列中的特定序列并对识别序列进行有效切割。
利用琼脂糖凝胶电泳,我们可以通过电泳结果检验DNA提取纯度、PCR和双酶切实验的效果,并对比各组实验效果了解实验各步骤的各项影响因素核试验操作。
2 实验材料和仪器
2.1实验材料
大肠杆菌DH5α(含重组了约1000bp DNA片断的PET32.a表达质粒,Amp抗性)
2.2 实验仪器
高压灭菌锅、超净工作台、恒温摇床、台式离心机、涡旋振动器、培养管、移液器、离心管、吸管头、量筒、紫外观测仪、小三角瓶、PCR仪、微量移液器、冰盒、制胶模、电泳仪。
3 实验试剂
3.1 DNA提取所需试剂
3mol/L醋酸钠(pH5.2)、TE缓冲液(pH8.0)、无水乙醇、75%乙醇、RNaseA(10mg/ml)、goldview DNA染色剂、氯仿、琼脂糖
溶液Ⅰ:50 mmol/L葡萄糖,25 mmol/L Tris-Cl(pH8.0),10 mmol/L EDTA (pH8.0);
溶液Ⅱ: 0.2 mol/L NaOH, 1%SDS(w/v),现配现用;
溶液Ⅲ:3 mol/L KAc,用冰醋酸调pH值至5.2;
3.2 PCR实验所需试剂
实验一提取的质粒DNA,0.5U/μL的 Taq酶及5 × PCR buffer、1 mmol/L 的dNTP、克隆基因特异引物PF、PR(各1µmol/L),0.2mL的PCR管,TIP头,goldview DNA染色剂,琼脂糖,10×上样缓冲液,0.5×TBE、marker DNA等。
PCR扩增引物:
PF:5`—CTCGGATCCATGGAGCTAGCTACTGCGGG—3`
PR:5`—CCGGTCGACTCCATCAATCTTGGAAAGCA—3`
3.3双酶切实验所需试剂
实验一提取的质粒DNA,BamHI(3 U /μL),HindⅢ(3 U /μL),1.5mL的EP管,TIP头,goldview DNA染色剂,琼脂糖,6×上样缓冲液,0.5×TBE等。
4 实验步骤
4.1 质粒DNA提取
4.1.1取1mL菌液放入Ep管中,12000 r/min(简写rpm)离心1min;弃尽上清;(注:先倒液体入废液缸,再倒扣用吸水纸吸干液体,残留液体可移液器吸干)
4.1.2加入200μL预冷的含RNaseA的溶液I,旋涡以充分混匀;
4.1.3加入200μL的溶液Ⅱ,温和混匀,室温静置5min以充分裂解细菌;
4.1.4加入200μL的溶液Ⅲ,温和混匀,-20℃放置8min(常温也可,但产量可能下降),12000rpm离心5min;吸上清至另一新的1.5mL EP管,加500μL氯仿/异戊醇(V:V=24:1)充分混匀,12000rpm离心5min;
4.1.5小心移取上清液500μL至另一新的1.5mL EP管,加入2倍体积无水乙醇,充分混匀,-20℃静置10min,12000 rpm离心10min,弃上清;(静置期间安排制备琼脂糖凝胶,每组制胶一块,具体见附件)加入200μL 70%乙醇洗DNA沉淀,12000rpm离心5min,倒掉乙醇,再12000rpm离心几秒钟,用移液器尽可能除去乙醇,烘箱适度风干;
4.1.6加入20μL RTE (含10μg/mL RNaseA 的TE,pH8.0)溶解质粒DNA, 37℃放置10min(时间充分可以考虑放置30分钟,以充分消化RNA)。
4.1.7取4~5μL质粒DNA溶液于另一干净离心管中,加入8μL TE及2μL上样缓冲液,混匀后全部点样于一个琼脂糖凝胶点样孔中。
4.1.8打开电源开关,调电压为80~100伏进行电泳 (注意电泳方向!!!)。
4.1.9电泳约40分钟后关掉电源,把凝胶置于紫外仪上观察结果并记录。
4.2 聚合酶链式反应(PCR)
4.2.1PCR反应液配制:反应体系为20µL (请先计算出如下各成分所需加入的量)
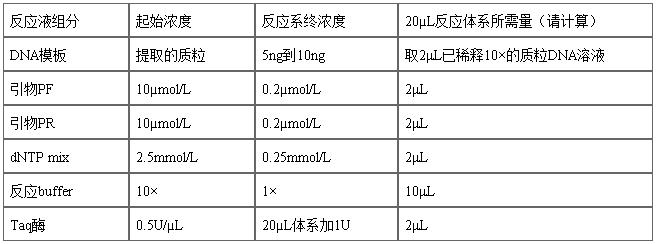
4.2.2把上述配制的反应液体系放置于PCR仪中,并使PCR仪运行如下PCR反应程序,实现靶DNA的扩增:
94℃ 3min→94℃ 50sec→54℃ 50sec→72℃ 1.5min→72℃ 5min
 ↑ 20循环 ↑
↑ 20循环 ↑
4.2.3 PCR扩增过程中,全班分组按实验一的方法制备1%的琼脂糖凝胶;
4.2.4 PCR结束后取10µL反应产物,加入2µL上样缓冲液,混匀并全部上样于1%琼脂糖凝胶1点样孔中;每块琼脂糖凝胶分别留一个点样孔并上样5µL marker DNA做标准分子量大小对照;
4.2.5接通电源,调电压80~90伏开始电泳,电泳持续约30~40分钟;
4.2.6切断电源,取出凝胶并置于紫外仪中观察、记录实验结果;
4.3 质粒DNA的双酶切分析
4.3.1在一1.5mL离心管中配制质粒双酶切反应液:(反应体系为20µL)
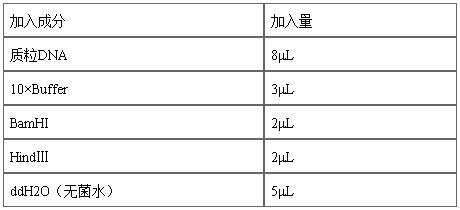
用移液器tip头轻轻混匀
4.3.2 37℃恒温条件下酶切反应约1-1.5h(酶切过程中,全班按实验一的方法分组各制备1%的琼脂糖凝胶);
4.3.3酶切结束后,往酶切反应管中加入4μL 6×上样缓冲液,轻轻混匀;
4.3.4 取10μL酶切液点样于琼脂糖凝胶1点样孔中;每块琼脂糖凝胶分别留一个点样孔并上样5µL标准分子量 marker DNA;
4.3.5接通电源,调电压80~90伏开始电泳,电泳持续约30~40分钟;
4.3.6切断电源,取出凝胶并置于紫外仪中观察、记录实验结果;
4.4 琼脂糖凝胶制备
4.4.1称量0.3g的琼脂糖粉置于一干净的小三角瓶中;
4.4.2加入30mL 0.5×TBE于三角瓶中,称出总重量并记录;
4.4.3在微波炉中充分溶解(约1分钟),称溶解后的总重量并用蒸馏水补充至原总重量(注意不要烫到手!!);
4.4.4室温条件下放置或自来水冲三角瓶下部到溶液温度冷却到约50度(即以手敢捏三角瓶为准,注意过程中要经常摇动防止降温不均匀导致局部凝固现象),此时再加入1.5μL goldview染料(原则上按照100mL琼脂糖液加goldview染料3-5μL),混匀;
4.4.5把混合液倒入一已插入梳子的制胶模具中,让其自然充分凝固(一般需要20分钟以上);
4.4.6小心拔掉梳子,把凝胶连同胶膜一起转移到电泳槽中(TBE缓冲液的添加以没过胶面1~2毫米为宜,注意电极方向),即可用于点样和电泳检测。
5 实验结果与分析
5.1质粒DNA提取所得凝胶电泳结果


由于实验操作不熟练,在添加上样缓冲液时不慎弄出了许多气泡,导致上样时基本吸不出来样品,从而上样量很少,最后电泳结果如图(左图)所示并不清晰
右图为别组实验结果。其实我组实验结果中1号样品还是能看到一点,仔细看能看到如右图4号样品相似的结果。
右图中4号样品有多条相近的条带,原因是提取的质粒DNA尽管相对质量相同,但由于各种因素导致会出现螺旋,线形和开环(开环质粒是指双链环状的质粒DNA有部分解链,因此电泳速度最慢)三种构像。经了解,三种构像的质粒在琼脂糖电泳的前后顺序是超螺旋>线形>开环,线形的质粒在中间,而开环的质粒在最后。
5.2 PCR扩增实验结果
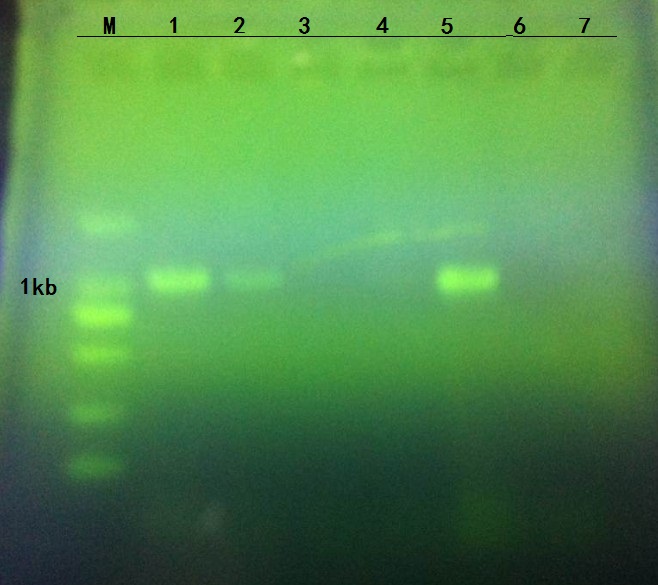
鉴于之前实验的经验,本次在添加上样缓冲液时有所注意并抓到了窍门,混匀后的样品基本没有气泡,但上样时依然遇到一些问题,图不少县戳破了凝胶,导致样品从凝胶下方漏掉了,结果图中3号、4号和6号样品没能出结果。图中1号、2号、5号样品在1kb位置有条带显示,表明目的序列得到了扩增,其中从显色效果可以看出,5号样品扩增结果最为理想。
据了解,PCR基本影响因素有变性-退火温度、引物设计、温度循环参数、不同的DNA聚合酶、模板序列本身。其中模板变性温度变性温度是决定PCR反应中双链DNA解链的温度,达不到变性温度就不会产生单链DNA模板,PCR也就不会启动。变性温度低则变性不完全,DNA双链会很快复性,因而减少产量。引物退火温度退火温度决定PCR特异性与产量;温度高特异性强,但过高则引物不能与模板牢固结合,DNA扩增效率下降;温度低产量高,但过低可造成引物与模板错配,非特异性产物增加。
另外引物设计影响PCR扩增的特异性,设计引物时通常会按照模板链长度决定引物长度,使其序列在改长度下仅可能出现一次。另外还要遵循引物内不含发夹结构等次级结构,引物间不互补。可按特殊需要设计特殊序列或增加其他功能集团。
不同的DNA聚合酶通常配合不同的目的载体,达到不同的功能与目的。如本次实验用的Taq酶具有在扩增序列3‘段结果添加一个腺嘌呤的功能,配合相关载体可达到扩增后便可用于载体构建的效果。另外不同酶的最大酶活力和最适催化条件也不一样,这也影响PCR结果。
最后模板序列本身对PCR也有影响,如模板序列中(G+C)%过大或过小、序列中存在次级结构等因素,这都会影响PCR扩增结果。
5.3质粒DNA的双酶切分析结果

理论上只会出现三种条带同时出现、二三种条带同时出现又或者只有第一种条带出现。另外仔细观察本次实验结果,发现Marker条带有点歪,这可能跟制胶质量有关。同时发现别组出现Marker走得十分歪,并且酶切样品一条条带都没有出现,这可能也跟制胶质量有关,然而经过多番讨论最后还是无法的出合理的结论。
本次实验还是有些样品从凝胶下方漏走了,导致1号和2号样品没有出现荧光条带。其中6号样品实验结果最为理想,在1kb位置明显出现了一条细细的荧光条带。在1kb位置出现的荧光条带为酶切所得的1kb目的序列,而上方荧光条带为切除了1kb目的序列后剩下的5kb的载体。据理解,实验可能会出现三种条带,一种是没被酶切或仅仅被切了一端的长度还是6kb的质粒DNA,该DNA特点是会再附近出现多种构象导致的三重条带;第二种是已被完整切去目的序列剩下的5kb的线性DNA;第三种是被酶切出来的1kb目的序列。
第二篇:华农差动放大电路_实验报告
实验五 差动放大电路
(本实验数据与数据处理由果冻提供,仅供参考,请勿传阅.谢谢~)
一、实验目的
1、加深对差动放大器性能及特点的理解
2、学习差动放大器主要性能指标的测试方法
二、实验原理
RP用来调节T1、T2管的静态工作点, Vi=0时, VO=0。RE为两管共用的发射极电阻, 它对差模信号无负反馈作用,不影响差模电压放大倍数,但对共模信号有较强的负反馈作用,可以有效抑制零漂。

差分放大器实验电路图
三、实验设备与器件
1、±12V直流电源 2、函数信号发生器
3、双踪示波器 4、交流毫伏表
5、直流电压表
6、晶体三极管3DG6×3, T1、T2管特性参数一致,或9011×3,电阻器、电容器若干。
四、实验内容
1、 典型差动放大器性能测试
开关K拨向左边构成典型差动放大器。
1) 测量静态工作点
①调节放大器零点
信号源不接入。将放大器输入端A、B与地短接,接通±12V直流电源,用直流电压表测量输出电压VO,调节调零电位器RP,使VO=0。
②测量静态工作点
再记下下表。
|
对地电压 |
Uc1 |
Uc2 |
Uc3 |
Ub1 |
Ub2 |
Ub3 |
Ue1 |
Ue2 |
Ue3 |
|
测量值 |
6.29 |
6.31 |
-0.74 |
-0.02 |
-0.02 |
-7.77 |
-0.61 |
-0.61 |
-8.39 |
2) 测量差模电压放大倍数(须调节直流电压源Ui1=0.1V ,Ui2=-0.1V)
3) 测量共模电压放大倍数
|
差模输入 |
共模输入 |
抑制比 |
|||||||||||
|
测量值 |
计算值 |
测量值 |
计算值 |
计算值 |
|||||||||
|
Uc1 |
Uc2 |
Uo双 |
Ad1 |
Ad2 |
Ad双 |
Uc1 |
Uc2 |
Uco双 |
Ac1 |
Ac2 |
Ac双 |
CMRR |
|
|
+0.1V |
10.08 |
2.55 |
7.46 |
6.29 |
6.31 |
-0.02 |
|||||||
|
-0.1V |
\ |
\ |
\ |
\ |
\ |
\ |
6.29 |
6.31 |
-0.02 |
||||
(理论上Uc1-Uc2=Uo双 ,由此可检验数据的准确性,但误差也是不可避免的)
理论计算:(r be=3K . 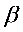 =100. Rp=330Ω)
=100. Rp=330Ω)
静态工作点:
 =
= mA
mA
IcQ=Ic3/2=0.577mA, IbQ=Ic/ =0.577/100=5.77uA
=0.577/100=5.77uA
UCEQ=Vcc-IcRc+UBEQ=12-0.577*10+0.7=6.93V
双端输出:(注:一般放大倍数A的下标d表示差模,下标c表示共模,注意分辨)
 =-33.71
=-33.71
Ac双 =0.
单端输出:
 =-16.86,
=-16.86, 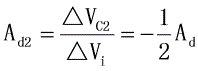 =16.86
=16.86
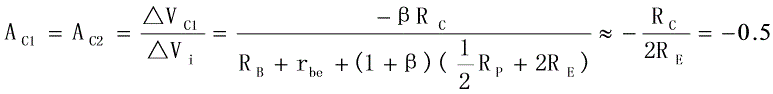
(参考答案中的Re=10K ,而Re等效为恒流源电阻,理想状态下无穷大,因此上式结果应为0.读者自己改一下)
实测计算:(注:本实验相对误差不做数据处理要求,下面给出的仅供参考比对数据)
静态工作点:
Ic1Q=(Vcc-Uc1)/Rc1=(12-6.29)/10mA=0.571mA Ic2Q=0.569mA
Ib1Q= IcQ/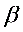 =0.571/100mA=5.71uA Ib2Q=5.69uA
=0.571/100mA=5.71uA Ib2Q=5.69uA
UC1E1Q=UC1-UE1=6.29-(-0.61)=6.90V UC2E2Q=6.92V
差模放大倍数:(Ui=Ui1-Ui2=+0.2V) (注:放大倍数在实测计算时,正负值因数据而异~!)
Ad1=(Uc1差模-Uc1)/(Ui-0)=(10.08-6.29)/(0.2-0)=18.95
Ad2=(Uc2差模-Uc2)/(Ui-0)=-18.80
Ad双=Uo双/Ui=7.46/0.2=37.3
相对误差计算 (||Ad理|-|Ad实||)/|Ad理|
rd1=|16.86-18.95|/16.86=12.4% rd2=|16.86-18.80|/16.86=10.9% rd双=10.6%
共模放大倍数:(Ui=+0.1V)
Ac1=(Uc1共模-Uc1)/Ui=(6.29-6.29)/0.1=0
Ac2=(Uc2共模-Uc2)/Ui=(6.31-6.31)/0.1=0
Ac双=Uc双/Ui=-0.02/0.1=-0.2 (Ui=-0.1V时同理)
共模抑制比:
CMRR=|Ad双/Ac双|=|37.3/(-0.2)|=186.5
4.单端输入(注:上面实验中差模与共模接法均为双端输入,详见最后分析)
|
电压值 |
放大倍数 |
|||
|
Uc1 |
Uc2 |
Uo |
||
|
直流+0.1V |
4.76 |
7.84 |
-3.70 |
|
|
直流-0.1V |
8.13 |
4.47 |
3.64 |
|
|
正弦信号 (50mV.1KHz) |
0.32 |
0.32 |
\ |
|
(正弦信号的Uc1=Uc2)
Ui=+0.1V时
Ac1=(4.76-6.29)/0.1=-15.3
Ac2=(7.84-6.31)/0.1=15.3
Ao=(-3.70/0.1)=-37.0
Ui=-0.1时
Ac1=(8.13-6.29)/(-0.1)=-18.4
Ac2=(4.47-6.31)/(-0.1)=18.4
Ao=3.64/(-0.1)=-36.4
正弦信号时(注:部分同学的输入电压可能为500mV,处理时请注意)
Ac1=(0.32-6.29)/0.05=-119.4
Ac2=(0.32-6.31)/0.05=-119.8
分析部分:(注:只供理解,不做报告要求)
Vi、Vo、Vc1和Vc2的相位关系
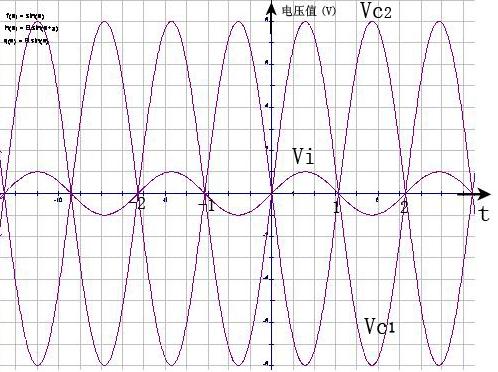
其中Vi、Vc1同相,Vi、Vc2反相,Vc1、Vc2反相。
Re的作用:Re作为T1和T2管的共用发射极电阻,对差模信号并无负反馈,但对共模有较强负反馈,可以有效抑制共模信号,即可以有效抑制零漂,稳定工作点。
恒流源(书上P165-166有介绍):恒流源作为负载时交流电阻很大,所以当用恒流源代替Re时,可以使差模电压增益由输出端决定 ,而和输入端无关。从数据中可以看到,用恒流源作负载时,抑制共模信号的能力大大提高了。

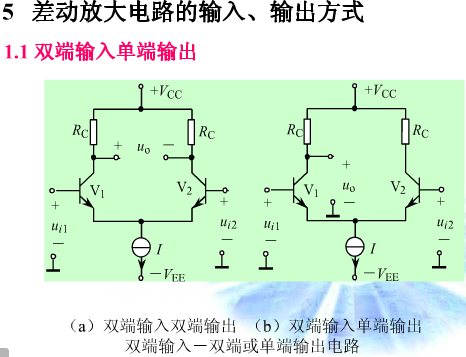
输入差模信号时Ui1=+0.1V , Ui2=-0.1V 输入共模信号时Ui1=Ui2=+0.1V或-0.1V
因为Ui1与Ui2均有信号输入,所以差模信号与共模信号属于双端输入
(输出方式则如图所示,只测一端电压Uoi为单端输出,测两端电压Uo则为双端输出)

单端输入时(注:Ui2此时接地)
接直流电压源Ui1=+0.1V或-0.1V 而Ui2=0
接交流电压源Ui1=50mV或500mV 而Ui2=0
即Ui1与Ui2只有一端输入信号,因此称为单端输入