电位滴定法测定氯化锂量
实验报告
天齐锂业股份有限公司 勾海霞
1 方法提要
试料以水溶解。在酸性溶液中,以银(或银─硫化银)电极为测量电极,甘汞电极为参比电极,以硝酸银标准滴定氯化物。用二级微商确定其反应终点,以消耗硝酸银标准滴定溶液的量计算氯化锂的含量。
2 试剂
2.1 硝酸(1+1),优级纯。
2.2 氯化钠标准溶液[C(NaCl)=0.1000mol/L]:称取5.8443g预先在450℃~500℃灼烧1.5h并在干燥器中冷却至室温的氯化钠(基准试剂),置于250mL烧杯中,加水溶解后,移入1000mL容量瓶中,以水稀释至刻度,摇匀。
2.3 硝酸银标准滴定溶液[C(AgNO3)=0.1000mol/L]。
2.3.1 配制:称取16.9872g硝酸银(分析纯),置于250mL烧杯中,加水溶解后,移入1000mL棕色容量瓶中,以水稀释至刻度,混匀。
2.3.2 标定:标定与试样的测定平行进行。
移取三份40.00mL氯化钠标准溶液(2.2),分别置于250mL烧杯中,加水至150mL,加入1滴溴酚蓝指示剂(2.4),加入1滴~2滴硝酸(2.1),至溶液恰呈蓝色,放入电磁搅拌子,将烧杯置于电磁搅拌器上,开动搅拌器,将测量电极(3.1.2)和参比电极(3.1.3)插入溶液中,连接电位计(3.1.1),调整电位器零点,记录起始电位值。
用硝酸银标准滴定溶液(2.3)进行电位滴定,其滴定方式为先加入10mL,再逐次加入一定量,快到终点时每次加入0.05mL,记录每次加入后硝酸银标准滴定溶液体积及相对的电位值E,计算出连续增加的电位值ΔE1和ΔE1之间的差值ΔE2, ΔE1的最大值即为滴定终点,到达终点后再记录两次电位值E。记录格式详见附录A(参考件).
滴定至终点所消耗的硝酸银标准滴定溶液(2.3)的体积V1按式(1)计算:
b
V1 =V2+V3×—— ………………………………… (1)
B
式中:V1——滴定氯化钠标准溶液(2.2)消耗硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
V2——电位增量值ΔE1达最大值前加入硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
V3——电位增量值ΔE1达最大值前最后一次加入硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
b——ΔE2最后一次正值;
B——ΔE2最后一次正值和第一次负值的绝对值之和。
平行标定所消耗硝酸银标准滴定溶液(2.3)体积的极差值不应超过0.10mL,取其平均值。
2.3.3硝酸银标准滴定溶液的实际浓度按式(2)计算:
C1×V4
C =─────── …………………………………(2)
V1
式中:C──硝酸银标准滴定溶液(2.3)的实际浓度,单位为摩尔每升(mol/L);
C1──氯化钠标准溶液(2.2)浓度,单位为摩尔每升(mol/L);
V4──移取氯化钠标准溶液(2.2)的体积,单位为毫升(mL);
V1──滴定氯化钠标准溶液(2.2)消耗硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
2.4 溴酚蓝指示剂(1g/L):用乙醇配制。
3 仪器
3.1 电位滴定装置
3.1.1 电位计:精度2mV。
3.1.2 测量电极:银电极或银—硫化银电极.
3.1.3 参比电极:双液接型饱和甘汞电极,滴定时外套管内装硝酸钾溶液(0.1mol/L) 。
4 分析步骤
4.1 试料
先称取试样4g左右,放入已恒重的带盖的称量瓶中,在250℃~260℃烘2h,于干燥器中冷至室温,称重。与称量瓶的质量之差即为试样质量,精确至0.0001g.。
独立地进行两次测定,取其平均值。
4.2 空白试验
随同试料做空白试验.
4.3测定
4.3.1 将试料(4.1)置于100mL烧杯中,用水溶解,移入250 mL容量瓶中, 以水稀释至刻度,混匀。
4.3.2 分取10.00 mL试液(4.3.1),置于250 mL烧杯中,加水至50 mL,加入1滴溴酚蓝指示剂(2.4),加入1滴~2滴硝酸(2.1),使溶液恰呈黄色,用硝酸银标准溶液(2.3)进行电位滴定.
注:滴定试液的速度与标定时的滴定速度应保持一致。
5 分析结果的计算
氯化锂的含量以氯化锂质量分数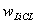 计,数值以%表示,按式(1)计算:
计,数值以%表示,按式(1)计算:
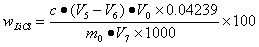 …………………….(3)
…………………….(3)
式中:
C——硝酸银标准滴定溶液(2.3)的实际浓度,单位为摩尔每升(mol/L);
V5——滴定试液消耗硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
V6——滴定空白溶液消耗硝酸银标准滴定溶液(2.3)的体积,单位为毫升(mL);
V0——试液总体积,单位为毫升(mL);
V7——分取试液体积,单位为毫升(mL);
m0——试料的质量,单位为克(g);
0.04239——与1.00 mL硝酸银标准滴定溶液[C(AgNO3)=0.1000mol/L]相当的氯化锂的质量,单位为克(g)。
6 实验方法
6.1加标回收实验
称样5.9947g氯化锂样品, 5.8451g NaCL(基准)稀释到1000mL。
分取5.00 mL试液,按分析步骤进行测定,结果见表1。
表1

结论
从实验结果中回收率可以看出结果有很高的可信度,从一系列的相关系数可以看出此方法精密度较高,从而得出结论:本方法可以满足氯化锂的含量测定。
6.2 pH值选择实验
以待测样品做此实验,称样4.0000克,按分析步骤进行测定,结果见表2。
表2
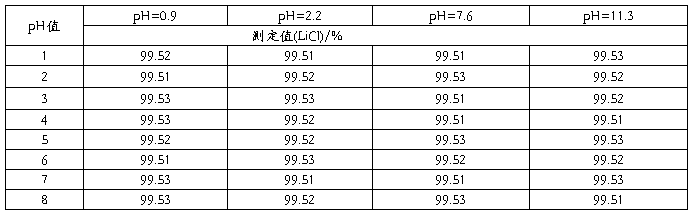
备注:此次实验中PH值对测定最后结果没有影响,但在实际测定时,pH为11.3的电位变化缓慢ΔE1值减小,初用此方法易对终点误判,而在酸性条件下突越变化明显,更易确定终点,而pH为2.2时最好控制,以溴酚蓝做指示剂,用1:1硝酸调至刚好黄色即可,故此方法选酸度为pH2-3为好。
6.3温度影响:
以待测样品做此实验,称样4.0000克,按分析步骤进行测定,结果见表3。
表3
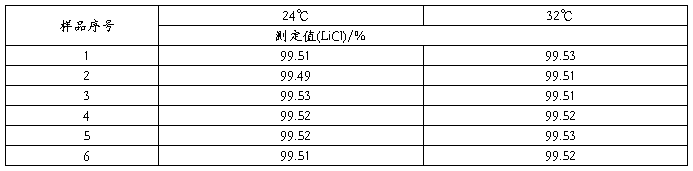
实验中此常规温度下对测定最后结果没有影响。
6.4干扰试验:
对比待测液加入2毫克硫酸根,按分析步骤进行测定,结果见表4。
表4
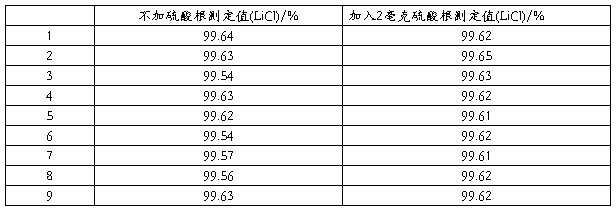
此试验中加入的硫酸根的量为样品所含量的100倍以上,由表可以看出,硫酸根在此量中不干扰测定。
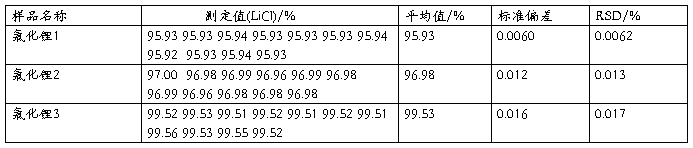
6.5 精密度
表5
6.6 结论
氯化锂离子选择电极法测定氯化锂结果准确、具有良好的稳定性,回收率达到99.8%以上。认为可以作为氯化锂测定的国家标准分析方法。
第二篇:三氯化六氨合钴的制备实验报告
一、实验目的
1. 掌握三氯化六氨合钴(III)的合成及其组成测定的操作方法, 通过对产品的合成和组分的测定,确定配合物的实验式和结构。
2. 练习三种滴定方法(酸碱滴定,氧化还原滴定,沉淀滴定)的操作。
3. 通过对溶液的配制和标定、仪器的使用、处理实验结果等提高学生独立分析能力、解决问题的综合能力。
二、实验内容——三氯化六氨合钴(III)的制备及组成的测定
Ⅰ、三氯化六氨合钴(III)的制备
(1)实验原理:
钴化合物有两个重要性质:第一,二价钴离子的盐较稳定;三价钴离子的盐一般是不稳定的,只能以固态或者配位化合物的形式存在。
显然,在制备三价钴氨配合物时,以较稳定的二价钴盐为原料,氨-氯化铵溶液为缓冲体系,先制成活性的二价钴配合物,然后以过氧化氢为氧化剂,将活性的二价钴氨配合物氧化为惰性的三价钴氨配合物。反应需加活性炭作催化剂。反应方程式:
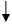 2CoCl2·6H2O + 10NH3 + 2NH4Cl + H2O2 ====2[Co(NH3)6]Cl3 + 14H2O
2CoCl2·6H2O + 10NH3 + 2NH4Cl + H2O2 ====2[Co(NH3)6]Cl3 + 14H2O
(橙黄色)
(2) 实验仪器及试剂:
仪器: 锥形瓶(250ml)、滴管、水浴加热装置、抽滤装置、温度计、蒸发皿、量筒(10ml、25ml、100ml)
药品:氯化铵固体、CoCl2·6H2O晶体、活性炭、浓氨水、5%H2O2、浓HCl、2mol/L的HCl溶液、乙醇溶液、冰、去离子水
(3)实验步骤:
在锥形瓶中,将4gNH4Cl溶于8.4mL水中,加热至沸(加速溶解并赶出O2),加入6g研细的CoCl2·6H2O晶体,溶解后,加0.4g活性炭(活性剂,需研细),摇动锥形瓶,使其混合均匀。用流水冷却后(防止后来加入的浓氨水挥发),加入13.5mL浓氨水,再冷却至283K以下(若温度过高H2O2溶液分解,降低反应速率,防止反应过于激烈),用滴管逐滴加入13.5mL5% H2O2溶液(氧化剂),水浴加热至323~333K,保持20min,并不断旋摇锥形瓶。然后用冰浴冷却至273K左右,吸滤,不必洗涤沉淀,直接把沉淀溶于50ml沸水中,水中含1.7ml浓盐酸(中和过量的氨)。趁热吸滤,慢慢加入6.7ml浓盐酸(同离子效应)于滤液中,即有大量橙黄色晶体([Co(NH3)6]Cl3)析出。用冰浴冷却后吸滤,晶体以冷的2ml 2mol/L HCl洗涤,再用少许乙醇洗涤,吸干。晶体在水浴上干燥,称量,计算产率。
Ⅱ、三氯化六氨合钴(III)组成的测定
(一)氨的测定
(1)实验原理:
由于[Co(NH3)]6Cl3在强碱强酸作用下,基本不被分解,只有在沸热的条件下,才被强碱分解,所以式样液加NaOH溶液作用,加热至沸使其分解,并整出氨,整出的氨用过量的2%磷酸溶液吸收,以甲基橙为指示剂,用HCl标准也滴定生成的磷酸氨,可计算出氨的百分量。
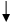 [Co(NH3)6]Cl3+3NaOH====Co(OH)3 +6 NH3++6 NaCl
[Co(NH3)6]Cl3+3NaOH====Co(OH)3 +6 NH3++6 NaCl
NH3+H3BO3 ==== NH4 H2BO3
NH4 H2BO3+HCl ==== H3BO3+ NH4Cl
(2)仪器与试剂:
仪器:250ml锥形瓶、量筒、pH试纸、滴定管
试剂:50ml2% H3BO3、HCl溶液、甲基红溴甲酚氯
(3)实验步骤:
(1)用电子天平准确称取约0.2g样品于250ml管中,加50ml去离子水溶解,另准备50ml2% H3BO3溶液于250ml锥形瓶中,
(2)在H3BO3溶液加入5-6滴甲基红溴甲酚氯指示剂,将样品溶液倒入加H3BO3的锥形瓶中,然后将锥形瓶固定在凯氏定氮仪上,开启凯氏定氮仪,氨气开始产生并被H3BO3溶液吸收,吸收过程中,H3BO3溶液颜色由浅绿色逐渐变为深黑色,当溶液体积达到100ml左右时,可认为氨气已被完全吸收(也可利用PH试纸检验氨气出口来确定氨气是否被完全蒸出)。
(3)用以用NaCO3溶液标定准确浓度的HCl溶液滴定吸收了氨气的H3BO3溶液,当溶液颜色由绿色变为浅红色时即为终点。读取并记录数据,计算氨的含量。
(二)钴的测定
(1)实验原理:
 利用三价钴离子的氧化性,通过碘量法,即利用I2的氧化性和I-的还原性进行滴定用来测定钴的含量,以淀粉作指示剂。主要反应方程式:
利用三价钴离子的氧化性,通过碘量法,即利用I2的氧化性和I-的还原性进行滴定用来测定钴的含量,以淀粉作指示剂。主要反应方程式:
[Co(NH3)6]Cl3+3NaOH====Co(OH)3 + 6NH3+ +6 NaCl
Co(OH)3+3HCl==== Co3++ 3H2O
2 Co3++2I-====2 Co2++I2
I2 +2S2O32- ====2I-+ S4O62-
⑵仪器与试剂:
仪器:250ml锥形瓶、250ml碘量瓶、电炉、量筒、pH试纸(精密)、电子天平、
试剂:KI固体、10% NaOH溶液、6mol/LHCl溶液、Na2S2O3溶液、2%淀粉溶液
⑶实验步骤:
用电子天平准确称取0.2g样品于250ml锥形瓶中,加入20ml去离子水,10ml 10% NaOH溶液,置于电炉微沸加热至无氨气放出(用PH试纸检验)。冷却至室温后加入20ml水,转移至碘量瓶中,再加入 1g KI固体,15ml 6mol/LHCl溶液,立即盖上碘量瓶瓶盖,充分摇荡后,在暗处反应10min后拿出。用已准确标定浓度的 Na2S2O3溶液滴定至浅黄色时,再加入1ml 2%的淀粉溶液,继续滴至溶液为粉红色即为反应终点(滴定开始阶段应迅速滴加,防止I2挥发)。读取并记录实验数据并计算钴的百分含量。
(三)氯的测定
⑴实验原理:
利用摩尔法测定氯的含量,即在中性或弱碱性溶液中,以K2CrO4作指示剂,用AgNO3标准溶液滴定Clˉ,由于2Ag++CrO42-=Ag2CrO4↓(砖红色), Ksp=2.0×10-12 ; Ag++Cl-=AgCl↓(白色), Ksp=1.8×10-10, 由于AgCl的溶解度比Ag CrO4小,根据分布沉淀原理,溶液中首先析出AgCl沉淀,化学计量点附近,由于Ag+浓度增加,与CrO42-生成砖红色Ag2CrO4沉淀指示滴定终点。另外为了准确滴定Cl-,需控制指示剂的浓度。根据实验数据计算氯的含量。
⑵仪器和试剂:
仪器:100ml容量瓶,250ml锥形瓶,酸式滴定管,玻璃棒,25ml移液管
试剂: 5%的K2CrO4溶液,AgNO3溶液
⑶实验步骤:
用电子天平准确称取约0.2g的样品用去离子水溶解,然后转移至100ml的容量瓶中定容,取25ml样品溶液于锥形瓶中,加入3滴5%的K2CrO4溶液作指示剂,用已准确标定浓度的AgNO3溶液滴定,溶液由黄色变为砖红色且砖红色30秒不消失(不需摇动)即为终点,读取并记录数据,计算氯的含量。
三、实验数据处理:
(1)三氯化六氨合钴的制备
[Co(NH3)6]Cl3的制备产率
得到产品m=0.72g
称取样品6.0g,理论产品质量为M=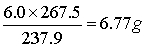
产率=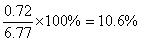

(2)氨的测定
氨的含量计算
m [Co(NH3)6]Cl3=0.2031g CHCl =0.3015mol/L
消耗15.56ml
根据反应方程式得HCl与NH3的计量比为 1:1
故样品中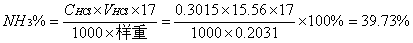
1mol样品中所含氨的物质的量为0.3973×267.5÷17=6.25mol
(3)钴的测定
已标定Na2S2O3的浓度为0.0150 mol/L
样品质量m=0.2001g
消耗Na2S2O3体积为54.95ml
根据化学反应方程式可知Co3+与Na2S2O3的计量比为1:1
故样品中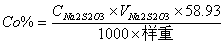
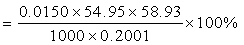
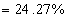
1mol样品中所含钴的物质的量为0.2427×267.5÷58.93=1.10mol
(4)氯的测定
称取样品的质量m样品=0.1998g
已标定的AgNO3的浓度CAgNO3=0.05844mol/L
滴定用AgNO3的体积VAgNO3=10.37ml
Ag++Cl-=AgCl↓ 计量比为1:1
所以  =
=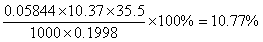
1mol样品中所含氯的物质的量为0.1077×267.5÷35.5=0.81mol

四、结果讨论误差与分析:
(1)样品产率低,原因有:
① 氯化铵加热时过度和温水浴时会释放出氨气,导致产率的下降。
② 冷水浴时不够完全,有部分固体没有析出或晶体析出后再溶解。
③ 抽滤时滤纸会粘附一部分,导致损失
④ 在反应搅拌与产品烘干时,都会导致损失
⑤ 加入的活性炭没有充分研磨,颗粒太大,催化效果差
⑥ 过滤时速度太慢
(2)通过对组分测定,氨、钴的含量偏高,氯的含量比较低。原因有:
① 氨的测定中,滴定终点判断有偏差,导致实验数据的误差。
② 钴的测定中,加淀粉时的黄色判断有点晚。
③ 钴的测定中,滴定管最后产生个小气泡,使Na2S2O3滴定的体积偏小,影响实验。
④ 氯的测定中,滴定终点不好把握,在没有达到终点时即停止滴定,导致误差较大。
⑤产品中可能有其他物质,比如二氯化五氨合钴。
⑥ 在读数时存在误差。
⑦ 配制标准溶液的过程中存在误差,导致滴定不准确,影响结果的计算。
五、注意事项:
(1)三氯化六氨合钴的制备:
① CoCl2·6H2O溶解后加入活性炭冷却不能太慢,因为氯化铵在溶液中加热后会有氨气放出,活性炭在使用前一定要充分研磨以提供较大的比表面积;
② 加H2O2前必须降温处理,一是防止其分解,二是使反应温和的进行。
③ 加H2O2时要逐滴加入,不可太快,因为溶液中的物质会与H2O2反应,会使反应太剧烈,会产生爆炸。
④ 两次冰浴冷却要充分,有助于沉淀的析出,提高产率。
⑤趁热吸滤后加入6.7mL浓HCl是用同离子效应增加产率,若浓HCl加入过多,会因稀释作用而产生盐效应而使溶解度加大,从而降低产率
(2)三氯化六氨合钴组分的测定:
① 分析天平称得质量要≥0.2g,因为分析天平的精确度为0.0001g,一次实验要称两次,误差为0.0002g,要求误差≤1‰,所以要大于0.2g;
② 滴定管快滴定完时,要把悬浮的液体刮锥形瓶,减小误差;
③碘量瓶要用磨口塞子,防止碘的升华;
④碘量法测定钴,在碘量瓶中加入KI固体和HCl后应立即将碘量瓶转移至暗处;
⑤Cl的测定中,在滴定后期,不要震荡锥形瓶,加入后产生砖红色,30秒不变色就为终点;
六、思考题:
1.在[Co(NH3)6]Cl3的制备过程中氯化铵,活性炭,过氧化氢各起什么作用
答.(1) 氯化铵的作用:
在没有铵盐的情况下,氨水遇钴盐后,即生成蓝色氢氧化钴(Ⅱ)沉淀:Co2+ + 2OH- →Co(OH)2↓。此沉淀易溶于过量的沉淀剂和铵盐溶液中。当有铵盐存在时,将抑制NH3·H2O的解离,即抑制OH-的产生使 [Co2+][OH-]2达不到 氢氧化钴(Ⅱ)的溶度积而形成[Co(NH3)6]2+,它随后被空气中的O2氧化,生成Co(Ⅲ)配合物。另外,氯化铵还能提供产物所需的NH3。
(2)活性炭起催化剂的作用,吸附反应物。
(3)过氧化氢起氧化剂的作用。
2. [Co(NH3)6]2+与[Co(NH3)6]3+比较,那个稳定,为什么?
答:[Co(NH3)6]3+稳定,因为[Co(NH3)6]2+中心原子采用sp3d 2杂化,即外轨成键,而[Co(NH3)6]3+中心原子采用d 2 sp3杂化,即内轨成键,根据配合物的价键理论,形成体与配位数相同的配合物,内轨型比外轨型稳定。
3、何为稀度?
答:所谓稀度即溶液的稀释程度,为物质的量浓度的倒数,如稀度为128,表示128L中含有1mol溶质。