仪器分析题库
第一章
1. 几个常用公式:
v=λν(v:传播速度,λ:波长,ν:频率)
1eV=1.602*10^-19 J
波动性:c=λf=f/σ(f:频率,σ=1/λ:波数)
微粒性:E=hν=hc/λ(h:普朗克常数,其值为6.626*10^-34 J·s)
2. 电磁波谱:电磁辐射按照波长或频率的大小顺序排列
γ射线→X射线→紫外光→可见光→红外光→微波→无线电波
(从左到右:波长越来越大,频率越来越小,能量越来越小)
3. 光谱法
按物质与能量作用形式(能量交换方向)分类:
1)吸收能量(基态→激发态)M+hν→M*
2)发射、辐射(激发态→基态)M*→M+hν
按作用的物质对象分类:1)原子光谱,2)分子光谱
4. 共振线:原子中的电子的基态和激发态能量差的辐射称为共振线
第一共振线:从基态跃迁至能量最低的激发态(第一激发态)产生的共振线称为第一共振线(由于各类元素的第一共振线不同,故这种共振线称为元素的特征谱线)。
第一共振线灵敏度最高,所以又称为最灵敏线。
第二章
1. 紫外-可见吸收光谱法(UV-Vis)是分子吸收光谱方法,也是带状光谱,是由分子中的价电子发生能级跃迁发生的。
2. 分子能级的高低顺序:σ<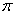 <n<
<n<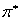 <σ*
<σ*
分子轨道间可能的跃迁有:σ→σ*, σ→, →σ*, n→σ*, →, n→
跃迁能量最大:σ→σ*,跃迁能量最小:n→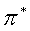
3. 朗伯-比尔定律
1)它表明在稀溶液中,物质对单色光的吸光度(A)与吸光物质溶液的浓度(c)和液层厚度(l)的乘积成正比。
2)公式:A= (
( :常数,称为吸光系数或吸收系数)
:常数,称为吸光系数或吸收系数)
3)摩尔吸光系数:在一定波长时,溶液浓度为单位摩尔浓度、液层厚度为单位厚度时的吸光度,其单位为L·cm-1·mol-1。
(偏离比尔定律的因素:1)化学因素:浓度,需要小于0.01mol/L;2)光学因素:非单色光;其他光学因素:反射,参比溶液;散射:胶体,细小颗粒物(应用均匀溶液,真溶液,若产生“假吸收”,会导致吸光度增加,导致结果偏高))
4. 影响显色反应的因素:显色剂用量、溶液酸度(pH)、显色时间、显色温度
第三章
1. 红外光谱(IR)是分子光谱,是由于分子中原子振动或分子转动产生的吸收光谱。
2. 红外光谱图纵坐标是百分透过率T或吸光度A,横坐标可用波长λ或波数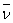 表示,波数即波长的倒数。
表示,波数即波长的倒数。
3. 红外吸收光谱产生的条件:
1)分子振动能级跃迁只能吸收与振动能级差的能量相当的辐射。(即辐射条件:振动吸收能量等于辐射产生的能量)
2)分子振动过程中必须有瞬间偶极矩的变化,这样的振动称为红外活性的振动
4. 常用术语:
1)特征峰:能用于鉴定官能团存在的吸收峰
2)相关峰:相互依存而又相互可以佐证的吸收峰
3)特征区:把波数在4000~1330cm-1(波长为2.5~7.5μm)的区间称为特征频率区,简称为特征区(主要包括-OH、-NH、-C=CH、-CH、-C=O、-C≡C、- C≡N、-C=C等伸缩振动)。
4)指纹区:波数在1330~667 cm-1(波长7.5~15μm)的区域,分子结构上的微笑变化,都会引起指纹区光谱的明显改变(有机化合物)
5. 几种化合物的红外吸收光谱:
1)饱和烷烃:C-H伸缩振动的波数为2950~2800(s)/cm
2)烯烃:=C-H伸缩振动的波数为3100~3010(m,w)/cm
3)炔烃:≡C-H伸缩振动的波数约为3300(s)/cm
第八章
1. 原子光谱法:测定金属及部分无机元素
1)原子吸收光谱法(AAS):一次侧一种物质
2)原子发射光谱法(AES):可同时侧十几种物质
3)原子荧光光谱法(AFS)
2. 原子吸收光谱法(AAS)又称为原子吸收分光光度法,其原理是基于蒸气中被测元素基态原子对其原子共振辐射的吸收强度来测定样品中被测元素含量的一种方法。
3. 影响吸收谱线宽度的因素:原子性质(自然宽度),外界影响(热变宽、压变宽、场变宽、自吸变宽等)
最主要的是多普勒展宽(即热变宽)
4. 原子吸光光度计:
构成:光源、原子化器、单色器、检测器
A. 光源:辐射待测元素的特征光谱,供测量使用。(提供的是锐线光源,产生窄带光谱或称锐线光谱;若光源为钨灯或氘灯发射连续光源,则灵敏度极差)
空心阴极灯:阴极(含待测元素)、阳极(钨、镍、钛等)、灯内充(惰性气体,如氖气、氩气等)
B. 原子化器:使样品溶液中的待测元素转化为基态原子蒸气
①火焰原子化器(原子吸收分光光度计)
雾化器:样品溶液→液滴→气溶胶
混合室:含有待测物质的气溶胶→与燃气助燃气充分混合
燃烧器:火焰中,蒸发、干燥、解离→基态原子
②非火焰原子化器(石墨炉原子吸收分光光度计)
电源:通电加热,2500K,试样会干燥、灰化、原子化
炉体:惰性气体
石墨管:样品管
C. 单色器:将灯发射的被测元素的共振线与其他波长的谱线分开(即分辨锐线光谱)
D. 检测器:将单色器分出的光信号进行光电转换
5. 原子吸收光谱中的干扰及抑制:
1)物理干扰
2)电离干扰
3)化学干扰(主要)分为阳离子干扰和阴离子干扰
原因:被测元素与共存的其他元素发生化学反应
结果:生成一种稳定化合物而影响原子化效率
消除方法:
①加入释放剂,使被测元素从化合物中释放出来
②加入络合保护剂,把被测元素保护起来
③加入助熔剂,如NH4Cl对很多元素具有消除干扰、提高灵敏度的作用(对高熔点的待测物起到助熔的作用)
④利用适当高温火焰消除干扰
⑤采用标准加入法
4)光谱干扰
第十一章
1. 固定相:固定在玻璃管内的填充物(固体或液体)
流动相:沿固定相流动的流体(气体或液体)
色谱柱:装有固定相的管子(玻璃或不锈钢制)
色谱法:在两相间进行多次分配而使混合物中各组分分离的方法
2. 色谱可分离和测定混合物,有机混合物常用GC(气相色谱:流动相为气体)或LC(液相色谱:流动相为液体),无机混合物常用IC(离子色谱)
3. 相关术语:
保留值:试样中各组分在色谱柱中的滞留时间的数值
1)保留时间(tR):被测组分从进样器开始到柱后出现浓度最大值时所需的时间
2)死时间(t0):不与固定相相互作用的物质,从进样开始到柱后出现最大值时所需的时间
3)调整保留时间(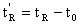 ):扣除死时间后的保留时间
):扣除死时间后的保留时间
4.分配系数:在一定温度和压力下,组分在固定相和流动相之间分配达平衡时浓度的比值

各组分的K必有差异,且K越大,混合物越易分离
5. 塔板理论:n=L/H=16 (块)
(块)
n:理论塔板数
L:柱长
H:理论塔板高的
tR:保留时间
w:峰底宽度
6. 定性分析:确定各色谱峰所代表的化合物(是什么物质),可用保留时间(即出峰时间)作定性指标。
7.定量分析:测定含量
mi=fi·Ai(mi:被测物系中组分i的质量或浓度,Ai:峰面积(或峰高hi))
fi:绝对校正因子,单位峰面积所代表物质的量
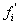 :相对校正因子,某物质与一标准物质的绝对校正因子的比值
:相对校正因子,某物质与一标准物质的绝对校正因子的比值
fi=mi/Ai,fs=ms/As(标准物质),
8. 定量分析常用方法:
1)归一化法: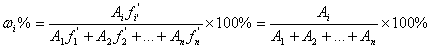 (同系物的相同)
(同系物的相同)
优点:简单,操作进样(密封状况)条件影响小,所有组分都分离
2)外标法
3)内标法
第十二章
气象色谱仪
1. 气路系统:提供流速稳定,纯净的载气(氮气、氢气、氦气、氩气)
2. 进样系统:进样器(微量进样器),汽化室(将液体样品瞬间气化)
3. 分离系统:色谱柱(先恒温,再程序升温:依据样品的沸点不同,在一个分析周期内柱温随时间由低温向高温作线性或非线性变化,以达到用最短时间获得最佳分离效果的目的)
4. 检测系统:检测器
1)热导检测器(TCD):通用型、广谱型(10^-9)
2)氢火焰离子化检测器(FID):测有机化合物,专属型,10^-12
原理:氢气在空气的助燃下经引燃后进行燃烧,以产生的高温为能源,使被测有机物组分电离成正、负离子,产生的离子在外电场的作用下定向运动而形成电流。微电流大小与进入离子室的被测组分的含量成正比,含量越高,产生的微电流越大。
3)电子俘获检测器(ECD):专属型,电负性强,测氯、氟
4)火焰光度检测器(FPD):专属型,测氮、磷
5. 记录系统:数据处理
6. 温控系统:汽化室、色谱柱、检测器温度
第十三章
1. 液相色谱法(LC)
高效液相色谱法(HPLC):优点在于高速、高效、高灵敏、高自动化
高速:由于使用高压输液设备,速度比经典液相色谱法快数百倍
高效:由于应用了颗粒极细(约5微米)、规则均匀的固定相,传质阻力小,分离效率高
高灵敏:由于配有高灵敏度检测器
2. 高效液相色谱仪:(高压输液泵、色谱柱、检测器—三大关键部件)
1)高压输液系统:储液器、高压泵、脱气器
2)进样系统
3)分离系统
4)检测系统
①紫外吸收检测器:仅适用于对紫外线(或可见光)有吸收的样品
优点:灵敏度较高,通用型较好
要求:试样必须有紫外吸收,但溶剂必须能透过所选波长的光,选择的波长不能低于溶剂的最低使用波长。
②荧光检测器:仅适用于发荧光的物质(稠环芳香烃、酶、氨基酸、维生素、色素、蛋白质)
它是选择性浓度型检测器,具有高灵敏度和高选择性
③示差折光检测器
④电导检测器
5)梯度淋洗:在分离过程中使流动相得组成随时间改变而改变。
作用相当于GC中的程序升温,可以改变流动相的组成,使性质改变,从而使分离效果得到改善
3. 正相键合相色谱法VS反相键合相色谱法
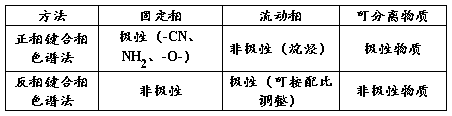
极性:分子中电荷分布的不均匀程度越不均匀,极性越大(相似相溶原理)
第十五章
1. 电分析化学:利用物质的电学及电化学性质来进行分析的一类方法
2. 电池表示方法:(-)电极a|溶液(a1)||溶液(a2)|电极b(+)
阳极 阴极
3. Nernst方程式
1)公式: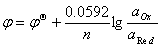 (n:电子转移数,a:活度)
(n:电子转移数,a:活度)
2)定义:计算电极上相对于标准电势来说的指定氧化还原对的平衡电压。
4. 参比电极:常用的有甘汞电极和银-氯化银电极
5. 金属指示电极:以金属为基体,基于电极上有电子交换反应的电极。包括第一类电极(活性金属电极)、第二类电极(金属-难溶盐电极)、第三类电极、零类电极
6. 离子选择性电极
1)pH玻璃膜电极:
原理:通过测定分隔开的玻璃电极和参比电极之间的电位差来测定
测定注意事项:
a) 使用前:电极放在蒸馏水或KCl溶液浸泡活化
b) 使用过程中:标准缓冲溶液校准:先用pH6.86(25℃)校准,然后用pH4.00(酸性)校准或pH9.18(碱性)校准
c) 测之前蒸馏水反复清洗电极,然后用吸水纸吸干
d) 溶液不能过酸或过碱,因为pH会影响测定过程线性关系
2)氟离子选择性电极:敏感膜由LaF3单晶切片制成
干扰及消除:
a) 酸度影响:过碱—释放F-,使测定结果偏高;过酸—生成HF,降低F-活度,使测定结果偏低。测定时,酸度应控制在pH5~7
b) 阳离子干扰:部分离子会与F-配合,使测定结果偏低。可加入配合掩蔽剂。
c) 基体干扰(以活度代替浓度):总离子强度调节剂(可同时控制pH、消除阳离子干扰、控制离子浓度)
第二篇:仪器分析各章知识点
各章知识要点
第2章 气相色谱分析
1.色谱法的分类(按两相状态)
2.何为GC法,GC定性定量的依据、定量方法及优缺点
3.GC分离原理(包括GSC法和GLC法)
4.气相色谱仪的构造
5.色谱流出曲线及其作用、色谱术语及换算关系
6.分配系数K和分配比k的定义、二者的异同点及相关计算
7.塔板理论的作用(包括H的n计算)
8.速率理论方程的作用(包括U最佳、Hmin的计算)
9.R的含义、作用
10.检测器的性能指标、四种检测器的适用特点及英文缩写
11.归一化法的使用条件、原理
12.内标法及内标物具备的条件
13.外标法的具体操作
第4章 电位分析法
1.电化学分析法、电位分析法、电位滴定法的定义。
2.电位分析法的测定依据。
3.电位测定法如何测定溶液的pH值(包括计算)。
4.指示电极、参比电极。
5.电位滴定法的原理及终点确定方法(重点掌握E/V曲线法和ΔE/ΔV—V 法及相关计算)。
6.电位滴定法的优点。
第5章 伏安分析法
1.极谱分析法及其特殊条件
2.极谱图及作用、极谱图上的各参数的定义及意义和作用
3.极谱分析定性定量的依据,半波电位的特性
4.极谱分析中的干扰及其消除方法
5.迁移电流
6、极谱分析的底液及其组成,各种物质的作用
7、极谱分析定量方法及其相关计算
8、单扫描极谱图的特征,单扫描极谱法定性、定量的依据(包括定性定量参数)
第8章 原子吸收光谱分析
1.AAS及基本原理
2.与其它光谱分析法相比,AAS的干扰少,具有相对高选择性。为什么?
3.何为共振线?在AAS中,是否一定以共振线为分析线?选择分析线的原则是什么?
4.在AAS中,被测物质是何微粒形式?
5.原子吸收分光光度计的基本组成部件有哪些?各部件的作用,常用何种光源?
6.何为光电倍增管的疲劳现象?如何防止或消除?
7.影响空心阴极灯发射特性的因素有哪些?关系如何?
8.在火焰原子化中,影响火焰温度的因素、火焰温度与原子化效率的关系?
9.AAS法定量的基础、定量方法及相关计算
10.AAS法适宜于常量分析还是微量分析?
11.AAS分析中,需控制哪些测定条件?
12.AAS分析中,常见的干扰有哪些?
13.何为化学干扰?有哪些具体形式?如何消除?
14.何为释放剂、保护剂、消电离剂?
15.何为原子分析中的灵敏度、特征浓度、检出限?它们与仪器的检测性能有何关系?
16.干扰形式的判断
a.在进行原子吸收分析,若在试样前处理时使用了硫酸或磷酸,从而导致其对测定元素的干扰,此干扰属于何种干扰形式?
b.待测元素与试样中共存元素的分析线重叠,引起什么干扰?
c.分析试液的粘度太大,使试液喷入火焰的速度不稳或降低,造成什么干扰?
第9章 紫外吸收光谱分析
1.UV法的概念
2.UV吸收光谱是怎样产生的?在UV光谱分析中,物质处于何种微粒状态?
3.按物质微粒形式,紫外光谱属何种光谱?若按产生机理,紫外光谱又称何种光谱?
4.分子内价电子及其跃迁类型;哪些跃迁产生的吸收光谱在紫外可见光区?紫外可见光区的波长范围?
5.助色团、生色团、红移、蓝移
6.K吸收带、R吸收带及它们的跃迁类型、强度。
7.紫外吸收光谱法的作用及其定性、定量的依据。
8.利用紫外吸收光谱推断物质的结构,其主要信息依据有哪些?
9.顺反异构体的UV光谱有何不同?
10.溶剂效应、影响该效应的因素及其关系。
11.紫外可见分光光度计的组成部件。
12.能够根据物质结构特征指出跃迁类型;由吸收光谱特征推断物质分子中的特征官能团。