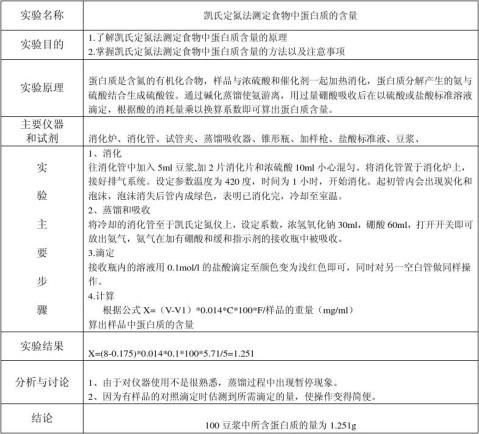
微量凯氏定氮法
姓名:XXX 年级 :20##级生物基地班 学号:201000140001
同组者:XXX 科目 :生物化学实验 实验时间:20##年6月3日
一、【实验目的】
1. 掌握凯氏(Kjeldahl)定氮法测定蛋白质含量的原理和方法
2.掌握微量凯氏定氮法的操作技术,包括标准硫酸铵含量的测定,未知样品的消化、蒸馏、滴定及其含氮量的计算等。
二、【实验原理】
天然有机物的含氮量常用微量凯氏定氮法来测定。生物材料的含氮化合物分析测定主要是指蛋白质,核酸的含量通常是用定磷法或别的方法测定。蛋白质的含氮量几乎是恒定的,约在15~16 %之间。因此只要测定蛋白氮,乘以6.25,即为粗蛋白质含量。
凯氏定氮法首先将含氮有机物与浓硫酸共热,经一系列的分解、碳化和氧化还原反应等复杂过程,最后有机氮转变为无机氮硫酸铵,这一过程称为有机物的消化。为了加速和完全有机物质的分解,缩短消化时间,在消化时通常加入硫酸钾、硫酸铜、过氧化氢等试剂,加入硫酸钾可以提高消化液的沸点而加快有机物分解,硫酸铜起催化剂的作用。使用时常加入少量过氧化氢作为氧化剂以加速有机物氧化。消化完成后,将消化液转入凯氏定氮仪反应室,加入过量的浓氢氧化钠,将NH4+转变成NH3,通过蒸馏把NH3驱入过量的硼酸溶液接受瓶内,硼酸接受氨后,形成四硼酸铵,然后用标准盐酸滴定,直到硼酸溶液恢复原来的氢离子浓度。滴定消耗的标准盐酸摩尔数即为NH3的摩尔数,通过计算即可得出总氮量。在滴定过程中,滴定终点采用甲基红-次甲基蓝混合指示剂颜色变化来判定。测定出的含氮量是样品的总氮量,其中包括有机氮和无机氮。以甘氨酸为例,其反应式如下:
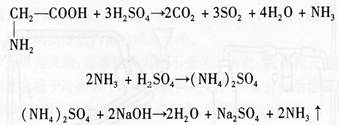
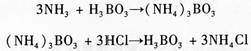
反应(1),(2)在凯氏烧瓶内完成,反应(3)凯氏蒸馏烧瓶中进行(图1)。
蛋白质是一类复杂的含氮化合物,每种蛋白质都有其恒定的含氮量(约在14%~18%,平均为16%)。凯氏定氮法测定出的含氮量,再乘以系数6.25,即为蛋白质含量。
三、【试验器材】
1. 牛奶
2. 凯氏定氮仪
3. 电炉
4. 消化架
5. 锥形瓶100ml(×5)
6. 量筒10ml(×1)
7. 滴定管(5ml,可读至0.02ml)
8. 凯氏烧瓶(×2)
9. 玻璃珠
10. 吸耳球
11.移液管(2ml,5ml,10ml×1)
四、【实验试剂】
1. 牛奶
2. 浓硫酸(A.R.)
3. 硫酸钾-硫酸铜混合物:硫酸钾3份与硫酸铜1份混合研磨成粉末。
4. 30%氢氧化钠溶液:30g氢氧化钠溶于蒸馏水,稀释至100ml。
5. 2%硼酸溶液:2g硼酸溶于蒸馏水,稀释至100ml。
6. 混合指示剂:0.1%甲基红乙醇溶液和0.1%甲烯蓝乙醇溶液按体积比4:1混合。
7. 0.00963mol/L标准盐酸溶液:用恒沸盐酸准确稀释标定。
五、【实验操作】
1.样品的消化
将两个50mL的凯氏定氮烧瓶编号(在烧瓶口附近),一只烧瓶内加1.0mL蒸馏水,作为空白。另一只烧瓶内加入1.0mL样液(牛奶液)。然后用取样器各加浓硫酸4mL(取浓硫酸时勿溅到衣物和皮肤上,也不要洒到实验桌上),用药勺加硫酸钾-硫酸铜混合物约200mg(不必称重,一点点即可),所有试剂要尽量加到凯氏定氮烧瓶的底部。烧瓶口插上小漏斗(作冷凝用),烧瓶置通风橱内的电炉上加热消化,注意先启动抽风机在消化开始时,应控制火力,不要使沸液冲到瓶颈。待瓶内水汽蒸完,硫酸开始分解并放出SO2白烟后,适当加强火力,继续消化,直至消化液呈透明蓝绿色为止。消化时间为2~3h,冷却,准备蒸馏。在消化时可以同时进行第二步。
2.定氮仪的洗涤
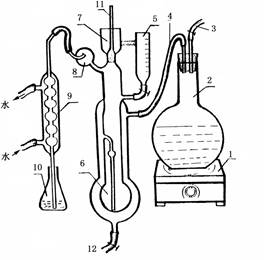
凯氏微量定氮蒸馏装置
1.电炉 2.蒸汽发生烧瓶 3.玻璃管 4.橡皮管 6.反应室 7.玻璃杯
8.气水分离器9.冷凝管10.锥形瓶11.棒状玻璃塞12.废液排出管
仪器应先经一般洗涤,再经水蒸气洗涤。
蒸气发生器中装加有H2SO4的蒸馏水和数粒沸石,加甲基橙指示剂后显粉红色。加热后,产生的蒸汽经贮液管、反应室至冷凝管,冷凝液体流入接受锥形瓶瓶。每次使用前,需用蒸汽洗涤5分钟左右(此时可用一小烧杯承接冷凝水)。将一只盛有5mL 2%硼酸液和1 ~ 2滴混合指示剂的锥形瓶置于冷凝管下端,使冷凝管管口插入液体中,继续蒸馏3分钟,如硼酸液颜色不变,表明仪器已洗净。若硼酸的颜色变为淡绿色,说明定氮仪内有残留氨,需要进一步用蒸汽洗涤。若反应室内有上次操作剩余的残夜,可以通过图1中7向反应式加冷的蒸馏水,然后短时间关闭4,则残夜会倒吸回贮液管,重复几次,并用蒸汽洗涤几分钟,再用上述方法检验是否已经洗干净。打开12废液排出管的夹子可以将废液放出。
3.标准品练习(标准硫酸铵溶液,含氮量0.3mg/ml)
仪器洗好后,取一100ml锥形瓶,加入5ml硼酸溶液,并使冷凝管下端玻璃管插入硼酸溶液中。取下11(图1)棒状玻璃塞,利用2ml移液管准确向反应室加入2ml硫酸铵溶液,然后将玻璃塞放回,向7(图1)玻璃杯中加入10ml30%NaOH溶液,旋转棒状玻璃塞,将氢氧化钠溶液缓慢地放入反应室中,并留少量液体作水封。等到锥形瓶内的硼酸溶液由紫红色变为鲜绿色后开始计时,继续蒸馏3min,然后移动锥形瓶使液面离开冷凝管口约1cm,继续蒸馏1min。并用少量蒸馏水洗涤冷凝口外围,移去锥形瓶。立即用标准盐酸溶液进行滴定,如果用滴定结果计算出的标准硫酸铵中氮含量接近于0.3mg/ml。则说明整个实验操作正确,可以进行下一步。
4.样品测定
仪器洗好后,取一100ml锥形瓶,加入5ml硼酸溶液,并使冷凝管下端玻璃管插入硼酸溶液中。取下11棒状玻璃塞,利用2ml移液管准确向反应室加入2ml消化好的样品溶液,然后将玻璃塞放回,向7玻璃杯中加入10ml30%NaOH溶液,旋转棒状玻璃塞,将氢氧化钠溶液缓慢地放入反应室中,并留少量液体作水封。等到锥形瓶内的硼酸溶液由紫红色变为鲜绿色后开始计时,继续蒸馏3min,然后移动锥形瓶使液面离开冷凝管口约1cm,继续蒸馏1min。并用少量蒸馏水洗涤冷凝口外围,移去锥形瓶。立即用标准盐酸溶液进行滴定,按上述方法洗涤仪器准备下一次蒸馏。重复蒸馏并滴定三次。
将2ml消化好的样品溶液改为2ml消化后的空白对照溶液,其他操作同上,测量三组。三组空白测量中,若锥形瓶中的硼酸溶液不变色,则无需滴定。
六、【注意事项】
1.凯氏法的优点是适用范围广,可用于动植物的各种组织,器官及食品等成组复杂样品的测定,只要细心操作都能得到精确的结果。其缺点是操作比较复杂,含有大量碱性氨基酸的蛋白质测定结果偏高。
2.普通实验室中的空气中常含有少量的氨,会影响结果,所以操作应在单独洁净的房间中进行,并尽可能快地对硼酸吸收液进行滴定。
3.定氮仪各连接处应使玻璃对玻璃外套橡皮管绝对不能漏气。蒸馏时需控制火力以避免样液倒吸。
4.消化时,若样品含糖高或含脂及较多时,注意控制加热温度,以免大量泡沫喷出凯氏烧瓶,造成样品损失。可加入少量辛醇或液体石蜡,或硅消泡剂减少泡沫产生。
5.消化时应注意旋转凯氏烧瓶,将附在瓶壁上的碳粒冲下,对样品彻底消化。若样品不易消化至澄清透明,可将凯氏烧瓶中溶液冷却,加入数滴过氧化氢后,再继续加热消化至完全。
6.蒸馏时,加入的氢氧化钠溶液除与硫酸铵作用外,还与消化液中的硫酸和硫酸铜作用。若加入的氢氧化钠不够,则溶液呈蓝色,不生成褐色的氢氧化铜沉淀。所以,加入的氢氧化 钠必须过量,并且动作还要迅速,以防止氨的流失。
7.蒸气发生瓶内的水装至2/3 体积并且保持酸性(在蒸气发生瓶内的水中加入稀硫酸,使之呈酸性,内加甲基橙指示剂数滴,水应呈橙红色,如变黄时,应该补加酸),以防止在碱性条件水中游离氨蒸出,使结果偏大。
8.因蒸馏时反应至外层的气压大于反应室内的压力,而反应室的压力大于大气压力,故可将氨带出。所以,蒸馏时,蒸气要均匀、充足,蒸馏中不得停火断气,否则,会发生倒吸。停止蒸馏时,由于反应室外层的压力突然降低,可使液体倒吸入反应室外层,所以,操作时,应先将冷凝管下端提高液面并清洗管口,再蒸1min 后关掉热源。
七、【实验结果】
1.结果
表一 各组实验消耗的标准盐酸体积(ml)
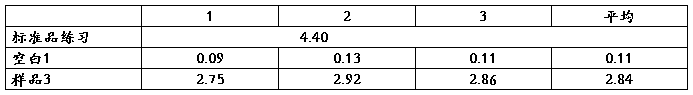
 2
.计算
2
.计算
 m =
m =
式中 m:样品中的含氮的质量,即100ml样品中含氮量(mg)
A:滴定样品消耗的HCl溶液体积(ml)
B:滴定空白消耗的HCl溶液体积(ml)
V:相当于未稀释样品的体积(ml)
c: 盐酸物质的量浓度(mol/L)
14.008:每摩尔氮原子质量(g/mol)
100:100ml样品
 所以可得:
所以可得:
 1. 标准硫酸铵中的N含量:m =
1. 标准硫酸铵中的N含量:m =
=0.297mg
该结果与标准值的相对误差=(0.297—0.3)/0.3×100% = 1%,这表示我们整个的实验操作正确。

 2
.蛋清中N含量(100ml样品):m =
2
.蛋清中N含量(100ml样品):m =
=18.41 mg
3.若样品中含氮物均为蛋白质,则蛋清样品中蛋白质的含量为:
18.41×6.25 = 115.063 mg
八、【实验结果分析】
1.在测量标准硫酸铵中含氮量时,我们测得的结果为0.297mg/ml,而标准值为0.3mg/ml,相对误差为1%,这说明我们组的实验仪器状态良好,而且操作也是正确规范的。
2.在测定样品中氮含量时,我们的测定结果为18.41mg N /ml,而且三组测量数据的相对偏差分别为2.55%,2.23%,0.76%,可见相互之间偏差很小,即实验准确性较高。由此推出实验室提供样品含氮量也应该在18.41mg N/ml左右。
九、【实验思考】
(一)本实验中所用各种试剂的作用。
1.浓硫酸:氧化剂,分解蛋白质成氨气和其他成分。
2.粉末硫酸钾-硫酸铜混合物(K2SO4: CuSO4·5H2O=3 : 1):充当催化剂,硫酸钾可以提高消化液的沸点,硫酸铜作为催化反应的主要成分。
3.30% NaOH溶液:碱化消化液,使硫酸铵分解释放出氨气,用于后续的滴定颜色反应。
4.0.010 mol/L盐酸:滴定蒸馏液,定量分析。
(二) 凯氏定氮法测定样品蛋白质含量时误差的主要来源以及应注意的事项。
凯氏定氮仪测定蛋白质含量的误差来源可能是样品、催化剂种类和用量、消化的时间、加碱液后的操作、蒸馏加热、清洗凯氏定氮仪的过程、凯氏定氮仪的气密性、氨气是否完全蒸馏出来;硼酸是否封住蒸馏管口、试剂的准确性、滴定终点的判断、滴定盐酸的浓度准确性等。
注意事项:
1、样品的取用量应适中,以免影响后续反应。
2、加入的催化剂硫酸钾和硫酸铜的配比适宜。
3、催化剂的添加量一定要准确,否则催化速度不一致,导致反应程度和挥发的氨不一样,误差大。
4、消化过程中,消化的时间应该充分,确保消化完全,消化至溶液呈现透明的浅蓝色。
5、消化后要将液体充分冷却,定容到容量瓶中备用。
6、消化之前检查凯氏定氮仪的气密性良好,后用蒸馏水清洗凯氏定氮仪,要充分清洗。
7、消化过程中,加碱液后要迅速关闭进样口,防止反应放出的氨气逸出。
8、消化过程中,火焰的温度要适当,太小加热过慢,太大容易导致溶液暴沸甚至从进气口溢出。
第二篇:凯氏定氮法实验报告
运动营养学实验报告
姓名:___ _____ 学号:_____ 日期:_______