分子生物学实验技术
(研究生、生物技术专业本科生必修课程)
课程简介:分子生物学实验技术已成为高水平医学科学研究中最常用的方法,本课程简要介绍有关基础知识,着重系统地进行有关操作技能的训练。
教学方式:通过实际操作,掌握有关知识和技能。
选课建议:本课程分四部分:DNA分析技术、RNA分析技术、PCR技术、蛋白质分析技术。请根据自己的研究方向所需技术手段选择其中三项内容。每部分人数限定72人(PCR技术不限人数)。
考核办法:实验报告+考试成绩
开设部门:生物科学系
总学时: 80
教学内容:
序号 主要内容 主讲人 实验员
一、DNA制备及分析技术
实验一、质粒的提取和纯化 徐建余 朱壮春
实验二、质粒的酶切、鉴定及片段的回收 周天戟 王艳茹等
二、RNA制备及分析技术
实验三、RNA的提取
实验四、RNA变性琼脂糖凝胶电泳
实验五、探针的标记
实验六、Northern杂交
三、PCR技术
实验七、聚合酶链反应
四、蛋白质分析技术
实验八、SDS-PAGE及分子量的测定
实验九、Western-Blot
实验十、凝胶图谱分析
“分子生物学实验技术”选课登记表
“分子生物学实验技术”选课登记表
实验一质粒DNA的提取和纯化
一、质粒DNA的提取
质粒:pBS[GAPDH]为4.0kb,其pBS载体2.8kb,GAPDH cDNA片段1.2kb。
宿主菌:E. coli HB101
试剂:
STE: 0.1mol/L NaCl
10mmol/L Tris·Cl (pH8.0)
1mmol/L EDTA
Sol I: (该液可配制100ml,67600 Pa即10磅灭菌15分钟,4°C储存)
50mmol/L Glucose
25mmol/L Tris·Cl (pH8.0)
10mmol/L EDTA
Sol II: (含0.2N NaOH、 1% SDS,新鲜配制)
10%SDS 4ml
1N NaOH 8ml
水 28ml
Sol III: (含3mol/L KAc、2mol/L CH3COOH, pH4.8)
5mol/L KAc 60ml
冰醋酸 11.5ml
水 28.5ml
碱裂解法小提质粒
1. 提取琼脂板上的单菌落,移至含有3ml LB培养液(含适当的抗生素)的培养三角瓶(20ml),37°C,强烈摇振过夜。(挑出3~5个菌落分别培养)
1. 取出培养液1ml移至Ep管中,4°C,12000g,30秒。
1. 弃上清,用1ml STE悬浮菌体沉淀,再离心回收菌体;再重复用1ml STE悬浮菌体沉淀,离心后,弃上清。
1. 将细菌沉淀悬浮于100ml冷Sol I中,强烈混匀。
1. 加200ml Sol II,颠倒离心管5次混匀悬液,冰浴3分钟(根据不同菌株可适当缩短时间:暴露在强碱性溶液中过长,共价闭合环状质粒DNA可发生不可逆的变性,导致内切酶切割困难,质粒DNA迁移率降低一倍左右,EB染色效率底)。
1. 加150ml Sol III,颠倒混匀10秒,冰浴3-5分钟。
1. 12000g,4°C离心5分钟,取上清移至新Ep管。
1. 加等体积酚/氯仿(1:1),振荡混匀;12000g,4°C离心2分钟。
1. 取上清至新管,加2倍体积95%乙醇(RT),振荡混匀,室温放置2分钟(不要-20°C沉淀,否则有较多盐析出)。
1. 12000g,4°C离心5分钟,弃上清。
1. 加1ml 70%冷乙醇,振荡漂洗沉淀。4°C 12000g 2分钟。
1. 弃上清,吸净管壁上的乙醇水珠,室温蒸发痕量乙醇。
1. 加入50ml TER(pH8.0)(TE中含无DNA酶的RNA酶20mg/ml)溶解DNA,短暂振荡5分钟后可进行内切酶酶切实验,或-20°C储存。
二、质粒DNA的纯化
聚乙二醇沉淀法
本方法经济简单,纯化的质粒可适用于细菌转化、酶切,尤其对减裂解法提取的质粒纯化效果更好。
1. 3ml质粒液移至一离心管加入3ml冷5mol/L LiCl,混匀,4°C,12000g,10分钟。
1. 将上清分装2管,加等量的异丙醇,混匀静置2分钟。室温12000g,10分钟。
1. 弃上清,流尽残夜,加70%乙醇漂洗沉淀及管壁。4°C,12000g,5分钟。弃上清,室温蒸发残存乙醇。
1. 加500ml TER(含无DNase的胰RNase 20mg/ml的TE,pH8.0)溶解沉淀,将溶液移至新Ep管,室温放置30分钟。
1. 加500ml 13%(w/v)聚乙二醇(PEG8000)的1.6mol/L NaCl,充分混匀,用12000g离心5分钟,以回收质粒。
1. 弃上清,用400ml TE(pH8.0)溶解沉淀质粒DNA。
1. 用酚、酚氯仿、氯仿各抽提一次。
1. 上清移至Ep管,加100ml的10mol/L乙酸铵,混匀,加2倍体积(约1ml)乙醇,混匀,室温10分钟。4°C, 12000g , 10分钟。
1. 弃上清(暂时保存上清,以防止不完全沉淀),200ml冷70%乙醇漂洗质粒DNA。4°C,12000g,2分钟。
1. 弃上清,蒸发残存乙醇。
1. 加500ml TE(pH8.0)溶解DNA(此步可适当减少TE量,以防浓度过稀),-20°C保存。
1. 测OD260、OD280值,进行DNA定量及纯度判断。
5ml样品+1ml水;OD260×10(mg/ml);1.6<OD260/280<1.8
实验二质粒的酶切鉴定及片段回收
一、小量酶解反应
主要应用于质粒的酶切鉴定。10ul反应体积中含1mg DNA,酶切反应体系如下:
质粒DNA 1mL
10×缓冲液 1mL
两种限制酶各 0.5mL +0.5mL
双蒸去离子水 7mL
总体积 10mL
[操作方法]
按下列顺序加入试剂:
(1)在一灭菌的新的Ep管中加入7ul双蒸水。
(2)加入10×缓冲液1mL 。
(3)加限制酶Kpn I 0.5mL,Sac I 0.5mL。
(4)最后加入质粒DNA lmL。
(5)稍离心,混合。
(6)37℃水浴1—1.5h。
(7)取出后电泳分析鉴定是否酶解。
[注意事项]
(1)双蒸水为可变体积,当其它反应成分确定后,用双蒸水将反应体积补足。
(2)质粒DNA最后加入,以防止操作不当引起试剂的污染。
(3) 大多数限制酶均加50%甘油缓冲液置于-20℃保存,其活力,稳定,但在配制反应混合物时,酶的加入量要准确限制小于总体积的l/10,否则甘油会抑制酶解反应。
二、冻溶法从琼脂糖凝胶中回收DNA
在重组DNA或探针标记等实验中,常常需要从凝胶中回收和纯化DNA,常用的回收方法有:压碎浸泡法、透析袋洗脱法、低熔点琼脂糖法和DEAE—纤维素膜法等。
[操作方法]
(1) 从琼脂糖凝胶中将含有拟回收DNA片段的胶块切下,置于Ep管内。
(2) 用一次性注射器将胶由针头处挤入Ep管内,加等体积Tris平衡酚混匀后,置液氮10min。
(3) 取出离心,5000g,5min。
(4) 小心地将水相转移至另一Ep管中。
(5) 加等体积酚:氯仿(1:1)充分混匀。离心,5000g,5min。
(6) 将水相转移Ep管中,加等体积氯仿:异戊醇(49:1)充分混匀。
(7) 离心,5000g,5min。
(8) 在转移出的水相中加1/10体积3mol乙酸钠(pH5.2)和2.5倍体积的预冷的无水乙醇置-30℃30min。
(9) 离心,12000g,15min.
(10)弃上清,加预冷的70%乙醇1ml,12000g,离心5min。弃上清,室温放置数分钟。
(11)将沉淀用20ul TE缓冲液(pH8.0)溶解,-20℃保存备用。
实验三真核细胞总RNA的制备
从细胞中分离RNA的纯度于完整性对于许多分子生物学实验至关重要。如Northern印迹与杂交分析、寡聚(dT)纤维素选择分离mRNA,cDNA合成及体外翻译等实验的成败,在很大程度上决定于RNA的质量。
RNA分离的最关键因素是尽量减少RNA酶的污染。
快速一步法提取总RNA
组织及有核细胞在匀浆过程中被变性液破膜、溶解,变性剂抑制RNA酶的活性,并使蛋白质变性及与核酸分离。经酸酚/氯仿将RNA抽提至水相,与DNA和蛋白质分离,再经异丙醇沉淀回收总RNA。
【试剂】
1×CSB缓冲液: 柠檬酸三钠 25mmol/L pH7.0
十二烷基肌苷酸钠 0.5%
b-巯基乙醇 0.1mol/L
配制100ml 5×CS缓冲液:柠檬酸三钠3.6762,十二烷基肌苷酸钠2.5g溶于100ml 0.05%DEPC水中,过滤,15lb/in2, 20min,去DEPC,4°C保存。
用前100ml工作液加 b-巯基乙醇700ml。
4mol/L异硫氰酸胍变性缓冲液:
47.26g异硫氰酸胍加至100ml CSB缓冲液中。
配制:23.63g异硫氰酸胍加热65°C溶于20ml无RNase水中,加10 ml 5×CS,加350ml b-巯基乙醇,用无RNase的水定量至50ml。(约加25ml水)
2mol/L 乙酸钠NaAc(pH4.0):
16.4g乙酸钠加至0.05%DEPC水40ml,用冰乙酸调pH至4.0,定容至100ml,分装,高压20min去除DEPC。
3mol/L 乙酸钠NaAc (pH5.2):
24.6g乙酸钠加至0.05%DEPC水80ml,用冰乙酸调pH至5.2,定容至100ml,分装,高压20分钟去除DEPC。
水饱和酚(酸酚,pH4.0):
取重蒸酚200ml,于65°C水浴溶解,加100ml无RNase水混匀,静置,取上层水相(大部分),加0.2g 8-巯基喹啉,混匀,保存于棕色广口瓶,4°C待用。
【操作方法】
1. 组织匀浆:新鲜的或液氮冻存的组织,称重后,按100mg组织/ml变性液,匀浆。置冰上30min。
培养细胞:贴壁细胞用PBS洗2次后,直接加2ml变性缓冲液/瓶;悬浮细胞用PBS洗沉淀2次,按107细胞/ml变性缓冲液,置冰上30min。
2. 取0.5ml粘稠液体加入1.5ml Eppendorf管(10ml塑料离心管)中,加50ml (1/10体积)2 mol/L NaAc(pH4.0),混匀。
3. 加0.5ml(等体积)酚和100ml (1/10体积的)氯仿:异戊醇(49:1),振荡混匀,冰浴10min。
4. 4°C,12000g,20min。
5. 吸上层水相入新管,加等体积异丙醇,-20°C,至少1hr。
6. 4°C,12000g,20min。
7. 弃上清,沉淀用1ml预冷的70%乙醇漂洗,并移入Eppendorf管。
8. 4°C,12000g,10min。
9. 弃上清,室温干燥蒸发残存乙醇,数分钟。
10.加100ml 水溶解RNA,测含量。
11.加1/10体积3mol乙酸钠(pH5.2)和2.5倍体积预冷的无水乙醇置-30℃30min。
12.离心,12000g,15min.
13.弃上清,加预冷的70%乙醇1ml,12000g,离心5min。弃上清,室温放置数分钟。
溶解RNA:每20mg RNA加4.5ml水溶解RNA,4℃存放待转膜(不宜久放)。
Total RNA Isolation
Guanidine-based isolation
Objective:
To obtain total RNA from zebrafish embryos.
Required Materials
· Denaturing Solution or Solution "D"
· 2 M NaOAC pH 4
· Phenol, H2O saturated
· 49:1 Chloroform/Isoamyl alcohol
· Isoproponal
· 75% EtOH
· DEPC-treated H2O or freshly deionized formamide
· 1 mL syringe
· 20 gauge needle
· 1.5mL microcentrifuge tubes
· microcentrifuge
Procedures
Start=> Collected zebrafish embryos of desired developmental stage, etc.
1. Remove excess H2O from embryos.
2. Using a 1mL syringe with a 20 gauge needle, add the appropriate amount (consult Table 1) of solution "D" to the embryos.
3. Homogenize the embryos by passage through the syringe needle (5-8) times. To decrease the volume of frothy homogenate and check the status of the embryos, briefly pulse in a microcentrifuge at low speed. If there is a pellet of embryonic tissue, continue to homogenate with the syringe needle. If not, continue with the extraction.
o The homogenates can be safely stored at -80°C.
4. Add 2M NaOAc pH4 (consult Table 1), and mix by inversion.
5. Add phenol and chloroform:isoamyl alcohol, vortex, and incubate on ice for 5-10'.
6. Centrifuge at 14,000 rpm for 2-3' at 4°C, and transfer upper aqueous phase to a new microcentrifuge tube.
7. Add 100% Isopropanol to precipitate RNA. Incubate samples at -20°C for at least 30'.
8. Centrifuge at 14,000 rpm for 10' at 4°C, and discard supernatant in waste container.
9. Dissolve the RNA pellet in solution "D", and transfer 10μL to a new microcentrifuge tube.
10.Precipitate RNA by adding equal volumes of 100% isopropanol. Incubate samples at -20°C for at least 30'.
11.Centrifuge at 14,000 rpm for 10' at 4°C, and discard supernatant in waste container.
12.Wash pellet by adding 75% EtOH.
o If RNA is to be stored, leave majority of RNA precipitated in the 75% EtOH at -80°C. Continue with the 10μL aliquot to determine approximate concentration and integrity of rRNA bands.
13.Centrifuge at 14,000 rpm for 10' at 4°C, and discard supernatant in waste container.
14.Pulse the pellet to collect remaining supernatant with a pipet tip. Allow the pellet to air dry or use a vacuum.
15.Resuspend in DEPC-treated H2O or formamide.
o Suspension in formamide protects the RNA from degradation by RNases, but for most applications the RNA should be precipitated from the formamide by adding 4 volumes of 100% EtOH and centrifuging at 14,000 rpm for 10' at 4°C.,
16.Quantitate RNA by diluting 1 μL into 100μL with alkaline H2O (see below). Then determine the A260 and A280.
o Water used for spectrophotometric measurement of RNA should have a pH of > 7.5. Acidic pH affects the UV absorption spectrum of RNA and significantly decreases the A260/A280. Adjust water to a slightly alkaline pH by adding concentrated Na2HPO4 solution to a final concentration of 1mM.
实验四 RNA甲醛变性凝胶电泳和转膜
从组织或细胞中提取出来的核酸(DNA/RNA)经琼脂糖凝胶电泳将其按分子量的大小分离,然后将其转移到固相支持物上(如:硝酸纤维素膜),用被标记的已知核酸片断(探针)对固相支持物上的待检测核酸进行检测(杂交)。
一、RNA电泳(甲醛变性电泳)
【目的】 将DNA/RNA通过凝胶电泳使之在凝胶中分离出来,通过加入标准核酸分子(markers)对其进行鉴定,并用于转膜、杂交等。
【试剂】
10×MOPS缓冲液:
0.2mol/L MOPS(3-〔N-吗啉〕丙磺酸)
80mmol/L NaAc
10mmol/L EDTANa2
先用400 ml DEPC水溶解20.6g MOPS和4.1gNaAc后,加入10ml 0.5mol/L EDTA,定容500ml。用0.22mm滤器过滤除菌,避光保存。
1×MOPS电泳缓冲液:
10×MOPS 150ml
37%甲醛 80ml
加水至1500ml。
甲醛凝胶变性RNA电泳加样缓冲液(10×):
50%甘油
1mmol/L EDTA
0.25%溴酚蓝
0.25%二甲苯青FF
5ml甘油、20ml 0.5mol/L EDTA、25mg溴酚蓝、25mg二甲苯青FF,加DEPC水至10ml,高压后,4°C保存。
5mol/L EDTA(pH8.0)
37%甲醛、甲酰胺等。
【操作步骤】
1. 电泳槽用0.3%H2O2浸泡30min,DEPC水冲洗晾干。
2. 铺胶(20ml)
琼脂糖 0.24g (1.2%)
无RNase水 17.4ml
10×MOPS 2.0ml
37%甲醛 0.6ml
EB 1ml
将琼脂糖加水融化后,加10×MOPS,待胶冷却至60°C左右,再加甲醛和EB。(若铺大胶可将上述各试剂的量加大2倍)。
3. RNA样品处理(以一条泳道为例)
10×MOPS 1.0ml
37%甲醛 3.5ml
甲酰胺 10.0ml
RNA样品 4.5ml(20mg RNA)
于65°变性15min,冰浴冷却。经此处理的RNA样品,-70°C可存放半年。
4.加2.0ml甲醛凝胶加样缓冲液(10×)
5.加样和电泳
将凝胶预电泳5min,电压降为5v/cm。随后加样品和标准物,以3-4v/cm电压降电泳,电泳液为1×MOPS电泳缓冲液。直至溴酚蓝迁移至胶下游的3/4处。
6. 电泳结束后,在紫外灯下,仔细测量28S rRNA和18S rRNA至加样孔的距离,并同荧光尺一起拍照,以记录标准物的位置。
【注意事项】
1. 每一泳道至多可分析30mg RNA,通常用10-20mg总RNA进行Northern杂交,可以检测高丰度mRNA(占mRNA总量的0.1%以上);如待测RNA含量极微,每个泳道加0.5-3.0mg poly(A)+ RNA。
2. 标准物28S rRNA»6333 base;18S rRNA»2366 base;9S兔b珠蛋白mRNA»710 base。 溴酚蓝在前(»300bp),二甲苯青FF在后(»4kb)。
二、RNA样品的转膜(虹吸印迹法)
【试剂】
20×SSC:175.3g NaCl,88.2g柠檬酸钠,DEPC水定容1000ml
NaOH调pH至7.0,高压后备用。
6×SSC: 用20×SSC稀释。
【操作步骤】
1. 将电泳后的凝胶用蒸流水漂洗2min,20×SSC浸泡30min。
2.剪一与凝胶大小相等的硝酸纤维素膜,用蒸流水浸湿后放入20×SSC中。
3.在方盘上放置一平板,其上放置一滤纸,滤纸两端搭入盘内浸入20×SSC中。
4.将凝胶倒置于平台上, 底面朝上,切掉右下角,并用保鲜膜封闭四周。
5.将硝酸纤维素膜放于胶上,赶走气泡。
6.膜上放两张滤纸,其上再放20层吸水纸。
7.吸水纸上放一平板,其上放500g左右的重物。
8.室温下过夜转膜,期间换纸2-3次。
9.完成转膜后,在紫外灯下观察效果,并标记好序号、加样孔位置和28S,18S的位置。
10.用6×SSC浸泡硝酸纤维素膜5分钟,滤纸吸干,80°C烤箱真空干燥2小时。室温保存。
实验五核酸分子探针的标记
随机引物法标记cDNA探针 随机引物是指含有各种可能排列顺序的寡聚核苷酸片断的混合物,因此它可以与任意核苷酸序列杂交,起到聚合酶反应的引物作用。
将待标记的DNA探针片断变性后与随机引物一起杂交,然后以此杂交的寡聚核苷酸为引物,在大肠杆菌DNA聚合酶I大断段(Klenow Fragment)催化下,合成与探针DNA互补的DNA链,当在反应体系中含有a-32P-dNTP时,即形成放射性同位素标记的DNA探针。
【试剂】 随机引物试剂盒(promega)
终止缓冲液: 0.2mol/L EDTA(pH8.0)
【操作步骤】
1. 探针100ng溶于8μl水中(1.5ml Eppendorf管),100 0C,5min变性(双链者打开双链),冰浴骤冷,离心片刻。
2. 标记程序: 终浓度
water μl
5× Labelling buffer 10µl 1×
mix dNTP (dATP dGTP dTTP) 2μl 20μΜ/each
变性的cDNA模板 25ng 500ng/ml
BSA (牛血清白蛋白) 2μl 400μg/ml
Klenow 5u 100u/ml
a-32P-dCTP 5μl (50µci) 333nM
总体积 50μl
3. 离心片刻,混匀。37 0C,1hr或室温3-12hr。
4. 100 0C ,2分钟,终止反应。冰浴,加1/10体积终止缓冲液,终止反应。直接使用或-20 0C保存或即刻纯化。
核酸探针的纯化
1.向上述Ep管中加入1倍体积的4mol/L乙酸胺(pH4.5)。加入2倍体积的预冷的乙醇,-200C,1hr。
2.4°C,12000g,5分钟,弃上清。
3.75%乙醇1ml漂洗, 4°C, 12000g 15分钟,弃上清。此过程重复2-3次。
4.加入100μl水溶解沉淀后,取1μl点在滤纸上,放入闪烁液3ml,测放射性比活度。
测量值×稀释倍数
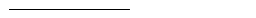
放射性比活度= cpm/mg DNA
探针浓度¸1000
理想的标记效果:放射性比活度大于108。
实验六 Northern杂交
[试剂] 终浓度
预杂交液:20×SSC 10 ml 5×SSC
50×Denhardt’s液 4ml 5×Denhardt’s液
10%SDS 2ml 0.5%SDS
10mg/ml变性断裂鲑精DNA 1ml 100-200µg/ml鲑精DNA
甲酰胺 20ml 50%甲酰胺
去离子双蒸水 3ml
预杂交和杂交
1. 将结合了RNA的NC膜浸泡于6×SSC液中,数分钟,使其完全湿润。
1. 将膜装在杂交管内,加适量的预杂交液(0.2ml/cm2)约15ml,尽量排除气泡,封口。然后置42ºC水浴摇床,轻摇8小时,可延长12-24小时。
1. 将已标记的双链DNA探针于95-100ºC加热5-10min,使之变性,然后迅速置冰浴中骤冷10分钟。如果是单链DNA或RNA探针,则不需要变性。
1. 将变性的标记探针加入预杂交液中,密封42ºC杂交过夜,可延长至24小时。
1. 洗膜:小心取出杂交好的膜,依次进行洗膜。
2×SSC, 0.5%SDS,室温, 5-10分钟
2×SSC, 0.1%SDS,RT, 10-15分钟
0.1×SSC, 0.5%SDS,37ºC, 30-60分钟
0.1×SSC, 0.1%SDS,65 ºC, 30-60分钟
1. 放射自显影:用0.1×SSC稍稍漂洗,吸干液体,放在洁净滤纸上,干燥,用保鲜膜包好,用X光片进行放射自显影, 加增感屏,-70ºC曝光7-10天。
实验七聚合酶链反应
【实验原理】
多聚酶链式反应(polymerase chain reaction, PCR)的原理类似于 DNA的天然复制过程。待扩增的DNA片段和与其两侧互补的两个寡核苷酸引物,经变性、退火和延伸若干个循环后,DNA扩增2n倍。
1.变性:加热使模板DNA在高温下(94℃)变性,双链间的氢键断裂而形成两条单链,即变性阶段。
2.退火:使溶液温度降至50—60℃,模板DNA与引物按碱基配对原则互补结合,即退火阶段。
3.延伸:溶液反应温度升至72℃,耐热DNA聚合酶以单链DNA为模板,在引物的引导下,利用反应混合物中的4种脱氧核苷三磷酸(dNTP),按5’®3’方向复制出互补 DNA,即引物的延伸阶段。
上述3步为一个循环,即高温变性、低温退火、中温延伸3个阶段。从理论上讲,每经过一个循环,样本中的DNA量应该增加一倍,新形成的链又可成为新一轮循环的模板,经过25~30个循环后 DNA可扩增106~109倍。
典型的PCR反应体系由如下组分组成:DNA模板、反应缓冲液、dNTP、MgCl2、两个合成的DNA引物、耐热Taq聚合酶。
【仪器】
1. PCR热循环仪
2. 琼脂糖凝胶电泳系统
【试剂】
1. 引物:根据待扩增DNA不同,所用引物也不同。选择引物时要符合一般引物设计的原则,必要时要用微机进行结构分析。
2. 耐热DNA聚合酶:此酶是从耐热细菌分离出来,能耐受93~100°C的高温而不失活。
3. 10×PCR缓冲液:
250~500 mmol/L KCl
100~500 mmol/L Tris-HCl pH8.4
15~20 mmol/L MgCl2
0.5% Tween-20
1 mg/L BSA(fiction V)无核酸酶
4.dNTPs储存液:
有商品化的中性混合液(2 mmol/L,10×) 供应,如需自己配制,则将dATP、dCTP、dGTP、dTTP钠盐各10 mg溶入2ml去离子水(浓度为5 mmol/L) ,用0.1mol NaOH调至pH7.0~7.5,分装,-20C°保存。
5.样品处理试剂:
不同样本所需试剂不同,根据具体情况而定。
【操作程序】
1. 提取样品模板;
2. 向一无菌微量离心管中依次加入:
ddH2O 补至终体积 100ml
10×PCR缓冲液 1/10体积
dNTPs 各 0.05 ~ 0.2mmol/L
引物 各 0.2 ~ 1 mmol/L
DNA模板 1~5ml (102~105拷贝)
Taq DNA Polymerase 1~2u
混匀后,离心数秒,加入液体石蜡100ml, 防止水分蒸发。
2. 95°C预变性5min;
1. 95°C变性45s~1min
2. 合适的温度复性45s~1min
3. 72°C延伸45s~1min
4. 3—5步重复进行25~35循环
5. 最后72°C延伸5~10min即完成PCR扩增
【产物的检测分析】
PCR产物可通过琼脂糖凝胶电泳检测,来判断产物的大小。
琼脂糖凝胶电泳常用试剂:
1. 电泳缓冲液:
TAE:Tris-醋酸缓冲液 或TBE:Tris-硼酸缓冲液
2. 上样缓冲液:一般为6×浓缩液
3. 溴乙锭(10mg/ml)
4. 1.5%~2%琼脂糖
淋球菌PCR
特异扩增淋球菌隐蔽性质粒DNA的Cpp B基因片断。
【试剂】
PCR混合液:
引物0.5mmol /L
dNTP各0.2mmol /L
10×buffer
2 mmol/L MgCl2
Taq DNA聚合酶
液体石蜡
上样缓冲液
裂解液:
10mmol/L Tris-HCl (pH 8.4)
10 mmol/L EDTA
0.2% Tween-20
100mg/ml蛋白酶K
2.5 mmol/L MgCl2
50mmol/L KCl
【试验操作】
1. 样品处理
取分泌物悬浮于1ml生理盐水中,14000rpm离心5min,弃上清,加50ml 裂解液悬浮沉淀,55℃水浴30min,95℃10min,14000rpm离心5min。
1. 扩增反应
在0.5ml无菌离心管中加入下列成分:
PCR混合液 20ml
模板(处理样品)2ml
液体石蜡25ml
混匀短暂离心,95℃变性5min,取出置冰上,加
Taq DNA聚合酶1ml
然后,按下面参数进行扩增:
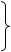
 94℃ (变性) 50s
94℃ (变性) 50s
55℃ (复性) 50s 35循环
72℃ (延伸) 50s
72℃ 最终延伸5min。
1. 扩增产物分析
向每管盖加2ml上样缓冲液,吸出20ml扩增产物,混匀,加样于2%琼脂糖凝胶,电泳后,在紫外灯下可观察到一条清晰的633bp的片段,即为阳性。
【注意事项】
1. 每管试剂在使用前应低速离心,使其中试剂沉于管底。
1. 加样顺序应合理化,避免交叉污染。
1. 预变性后应立即进行扩增,避免室温放置。
1. Taq DNA聚合酶必需-20℃保存。
实验八 SDS-PAGE测蛋白质的分子量
采用十二烷基硫酸钠-聚丙稀酰胺凝胶电泳(SDS-PAGE,polyacrylamide gel electrophoresis)方法可对蛋白质的组分进行分离,并可精确测得蛋白质的分子量。常用的方法为SDS-PAGE不连续系统。
【基本原理】聚丙稀酰胺是由丙稀酰胺(acrylamide)和N,N’-亚甲基双丙稀酰胺(N,N’-methylene bis acrylamide)经共聚合而成。此聚合过程是由四甲基乙二胺(tetramethylethylenediamine,TEMED)和过硫酸胺(ammonium persulfate,AP)激发的。被激活的单体和未被激活的单体开始了多聚链的延伸,正在延伸的多聚链也可以随机地接上双丙稀酰胺,使多聚链交叉互连成为网状立体结构,最终多聚链聚合成凝胶状。
不连续系统由上层浓缩胶和下层的分离胶组成。浓缩胶(pH6.7,孔径大)主要作用是使样品浓缩,使样品在未进入分离胶前,被浓缩成很窄的条带,从而提高分离效果。分离胶(pH8.9,孔径小)通过分子筛效应和电荷效应,把样品中的各组分按分子量和电荷的大小而分开。
如果要利用凝胶电泳测定某一蛋白质的分子量就必须将电荷效应去掉或减少到可以忽略不计的程度,使蛋白质泳动率的大小完全取决于分子量。如何去除电荷效应呢?现常用的是十二烷基硫酸钠(SDS)。SDS是一种阴离子去污剂。在电泳体系中加入一定浓度的SDS,SDS以一定的比例和蛋白质分子结合成复合物,使蛋白质分子带负电荷,这种负电荷远远超过了蛋白质分子原有的电荷,从而减低或消除了各种蛋白质分子天然电荷的差异。
【试剂】
Sol A:30%凝胶
丙烯酰胺30g,N,N’-亚甲基丙烯酰胺0.8g,加水至100ml。棕色瓶4°C避光保存。
Sol B:分离胶缓冲液(1.5mol/L Tris-Cl, pH8.9)
Tris 36.3g,1N HCl 48ml,补水至200ml。
Sol C:浓缩胶缓冲液(1mol/L Tris-Cl, pH6.7)
Tris12.1g,1.6N HCl 50ml,补水至100ml。
10% SDS
10% AP(过硫酸铵,ammonium persulfate)现用现配
TEMED(N,N,N,N’-四甲基乙二胺)(v/v)
1% Agar: Agar 0.3g
Sol B 7.5ml
10% SDS 0.3ml
水 22.2ml
电泳缓冲液: Tris 15g
Glycine 72g
10% SDS 50ml
加水至5升(pH8.5)
样品缓冲液(2×): 10%甘油
2%SDS
5%b-巯基乙醇
20mmol Tris-Cl, pH6.7
0.05%溴酚蓝
棕色瓶4°C避光保存。
染色液:0.25%(w/v)考马斯亮蓝R-250
考马斯亮蓝R-250 1.25g
50%甲醇 450ml
冰醋酸 50ml
脱色液: 冰醋酸 75ml
甲醇 50ml
水 875ml
异丁醇
标准分子量的蛋白质
【操作方法】
1.安装垂直板电泳槽
先将长短不等的两块玻璃板洗净,浸泡乙醇中。用前取出晾干,嵌入∪垂直板胶带模凹槽内。长玻璃板下沿与胶带框底之间保留2-3mm距离,使此端的凝胶与阳极相通;而短玻璃板的下沿则插入橡胶模的底槽内,三边均密封。
按“夹芯”法将上述凝胶模子装入电泳槽内。注意电极方向,短玻璃板位于阴电极槽一侧(白金电极丝在槽的上方),长玻璃板位于阳极一侧(白金丝在槽的底部)。
2. 用滴管吸取融化的1%琼脂糖溶液,灌入凝胶模板底部(长玻璃板外侧底部),封住底部的窄缝(通向阳极的盐桥)。
3. 配胶 根据所测蛋白分子量范围,选择适当的分离胶浓度,按表所列的试剂用量和加样顺序配制胶。表中为30ml分离胶和10ml浓缩胶,可按实际用量来按比例增加或减少上述试剂。
SDS-不连续系统不同浓度凝胶配制用量表

分离胶: 7% 10% 12% 20% 3%浓缩胶

Sol A 7 10 12 20 1.0
Sol B 7.5 7.5 7.5 7.5 Sol C 1.25
10%SDS 0.3 0.3 0.3 0.3 0.1
TEMED 0.02 0.02 0.02 0.02 0.02
双蒸水 15 12 10 2 5.55
混匀,抽气去氧
10%AP 0.2 0.2 0.2 0.2 0.1

4. 灌注分离胶 一旦加入AP后,轻轻混匀,立即小心地将分离胶灌入准备好的凝胶模子玻璃板间隙中,上端保留3cm高的空隙。用吸管将异丁醇覆盖分离胶液面(压平分离胶面;阻止空气中的氧对凝胶聚合的抑制)。约30分钟聚合完成(形成明显的界面),倒去覆盖液,用1ml Sol C涮洗2次,用吸水纸尽可能地吸干残液。
5. 灌注浓缩胶 用吸管将浓缩胶加在分离胶上,插入合适的样品孔梳子,避免出现气泡。室温下聚合约30分钟,细心拔出梳子,用双蒸水涮洗样品孔,去除未聚合的丙烯酰胺,用滤纸吸干残液。
6. 处理样品 将样品(蛋白质浓度以1-1.5mg/ml浓度为宜)与等量样品缓冲液混匀,100°C,3分钟。标准分子量蛋白质,按1mg/ml比例溶解,取出适当的标准品加等量的样品缓冲液混匀,煮沸2分钟,取出冷却。将处理好的样品与标准品,以每孔20-50ml(根据样品孔的大小及蛋白质的浓度决定),用微量移液器轻轻加入样品孔内。
7. 电泳 加样后,在电泳槽两侧加电泳缓冲液。电泳液面要高于样品孔(小心加电泳液,使其慢慢地漫过样品孔以免扰乱样品)。接好电极(注意正负极方向)电泳,开始电压为8v/cm凝胶,待染料进入分离胶后,将电压增加到15v/cm胶,继续电泳直至染料抵达分离胶底部,断开电源。
8. 取下凝胶,固定,染色,脱色;或进行Western blot分析。
【注意事项】
1. 丙烯酰胺有神经毒性,可经皮肤、呼吸道吸收,故操作时注意防护。
2. 加样的量要适当,0.25mg某种蛋白质,即可观察到其电泳带;如果有20-100mg,便过量。
凝胶考马斯亮蓝染色
1. 电泳结束后,用至少5倍体积的染色液浸泡凝胶,放在摇床上室温缓慢旋转3-4小时。
2.回收溶液,用脱色液浸泡凝胶,缓慢摇动4-8小时脱色,其间换液2-3次,直至满意为止。
3.将脱色后的凝胶照相或干燥,也可用塑料袋密封在20%甘油水溶液中长期保存。
此方法检测的灵敏度为0.2~1.0mg。
分子量计算
根据标准蛋白质分子量的对数和泳动率呈线性关系而求出未知蛋白质的分子量。

 泳动率=
泳动率=
1.计算每个蛋白条带的泳动率 距离:从加样孔始测量到各个条带。
2.根据标准蛋白条带计算出线性方程或画出标准线。
3.代入未知蛋白条带的泳动率,计算器分子量。
实验九 WESTERN BLOT检测蛋白
与DNA的Southern blot印迹相对应,蛋白质分析中应用的Western blot印迹技术也是通过对目标组分电泳分离,并转移至固相支持物(硝酸纤维膜),利用特异抗体作为探针,对靶物质进行检测。
蛋白质的Western blot结合了凝胶电泳的高分辨率和固相免疫检测的特异敏感等多种优点,可检测到低至1-5ng中等大小的靶蛋白。
【试剂】
转移缓冲液: 25mM Tris-Cl, 192mM甘氨酸,20%甲醇(v/v), 0.01%SDS, pH8.3
漂洗液: PBS-Tween 20
150mM NaCl, 10mM Tris-Cl(pH7.4), 0.05% Teeen-20(v/v)
封闭液: 5%脱脂奶粉或3%BSA溶于10mM PBS-Tween 20
一抗: 特异的单克隆或多克隆抗体
酶标二抗
底物液:5mg DAB/ml 溶于0.05M柠檬酸-磷酸盐缓冲液(pH5.0)
【操作方法】
1.剪6张与电泳凝胶大小一致的Watman 3MM滤纸和1张硝酸纤维膜(NC),带手套操作。
2.将上述滤纸及NC膜,浸入双蒸水中,再浸泡转移液中。
3.凝胶用双蒸水漂洗2次,再用转移液漂洗10分钟,2次。
4.按图示装好凝胶与NC膜,注意NC膜的药膜面朝向凝胶,膜与凝胶之间无气泡。
5.将装好的胶板插入转移槽中,注意NC膜位于阳极侧。
6.槽中加转移液,使之漫过铂金丝,接好冷却水系统。
7.电泳:6-10v/cm (电极距离),1-16小时(依据转移效果)。
8.电泳完毕后,取出NC膜,用铅笔做好标记,漂洗液洗3次,每次5分钟。
9.封闭: 将NC浸入封闭液中1小时或4℃过夜。用漂洗液洗3次,每次5分钟。
10.将NC膜切成小条,放入反应槽中,加入稀释好的一抗,同时做阴阳对照。室温反应2小时,或4℃过夜。振荡漂洗3次,每次10分钟。
11. 加酶标二抗,室温反应1-2小时,振荡漂洗3次,每次10分钟。
12. 显色:加入底物液,至蛋白带清晰为止。
13. 终止反应: 水漂洗。
【转移事项】
1. 电泳毕可用含0.2%丽春红S的3%冰醋酸水溶液染NC膜,来观察转移结果和标准蛋白位置。 或用考马斯亮蓝染凝胶来观察转移效果。
2. 酶标二抗可以是IgG, IgM, IgA. 也可用酶标的SPA蛋白。
3.转移好的NC膜,如不接着做酶标反应,可在封闭后,漂洗,晾干,封于塑料袋中,4℃保存备用。