ָ�����и������У���ϵ��?U��?H��?S��?A��?G�к���Ϊ�㣿
�� �����������������̣�
�� ʵ��������������̣�
�� ����������(p1,T1)״̬���ȿ���仯��(p2,T2)״̬��
�� H2��Cl2�ڸ��Ծ��ȵ������з�Ӧ����HCl��
�� 0�桢 ʱ��ˮ��ɱ��������̣�
ʱ��ˮ��ɱ��������̣�
�� �������忨ŵѭ����
(1) ��U = ��H = 0�� (2) ��H = 0�� (3) ��S = 0�� (4) ��U = 0��
(5) ��G = 0�� (6) ��U����H����S����A����G��Ϊ 0��
�زS���ǹ��̵������̡�7. ×; �زS�ǿ�����̵�������.
�ڹ���ϵͳ�з������κι��̶����Է����̡���
������ȹ��̱ض��ǵ��ع��̡���
��ŵ�Ȼ���Ч��ֻ��������Դ���¶��йض��빤�������ء���
���������һ�����Է����̡�×���������Ҳ�з��Է����̡�
ָ�����й�ʽ����������
��1��dU = ��Q �C PdV 1. �����ϵ������Ϊ0
��2����H = QP�� ��U = QV 2. ��H=QP, �����ϵ��ƽ��̬����������������ѹ����
��U=QV, �����ϵ��ƽ��̬�����������������ݹ���
��3��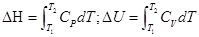
3. 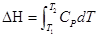 �������ϵ��ƽ��̬��״̬�����仯�ĵ�ѹ����
�������ϵ��ƽ��̬��״̬�����仯�ĵ�ѹ����
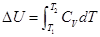 �������ϵ��ƽ��̬��״̬�����仯�����ݹ���
�������ϵ��ƽ��̬��״̬�����仯�����ݹ���
�����������壬������һ�й��̡�
��4��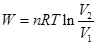 4. �����ϵ��ƽ��̬������������������������¿�����̡�
4. �����ϵ��ƽ��̬������������������������¿�����̡�
��5��W = ��P��V 5. �����ϵ��ƽ��̬������������������ѹ������
��6��PV�� �� ������P1-�� T���� ������TV�� -1�� ����
(��=CP/CV, ��ԭ��Cv=3R/2;Cp=5R/2,˫ԭ�� Cv=5R/2;Cp=7R/2,��
6. �����ϵ��ƽ��̬������������������������ȿ�����̡�
��7��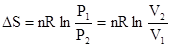
7. �����ϵ��ƽ��̬���������壬���¹��̣�������������
��8��
8. �����ϵ��ƽ��̬��״̬�����仯�����¹��̣�������������
��9��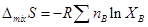
9�������ϵ��ƽ��̬����������,������Һ���µ�ѹ��ϡ�
.��2-12 ����1.00molH2O(1��25°C��101kPa)���µ�ѹ�������̵�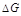 �����жϴ˹����Ƿ��Է����С���֪H2O(1)��25°Cʱ�ı�������ѹΪ3168Pa��
�����жϴ˹����Ƿ��Է����С���֪H2O(1)��25°Cʱ�ı�������ѹΪ3168Pa��
�⣺��������п�����̣�
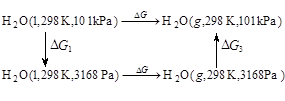
������ 
�� ��Һ���ڸı�ѹ��ʱ��
��Һ���ڸı�ѹ��ʱ�� ��֮�����ڸı�ѹ��ʱ��
��֮�����ڸı�ѹ��ʱ�� С�ܶ࣬���Ժ���С�ơ�
С�ܶ࣬���Ժ���С�ơ�
���

���ڵ��µ�ѹ��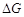 >0���ʴ˹��̲����Է����С�
>0���ʴ˹��̲����Է����С�
P51 ����1-10 ���庤��0�棬5×105Pa��10dm3��ʼ̬������һ���ȿ������������105Pa���Լ�����̬���¶�Ϊ���ɣ��˹��̵�Q��W�� ��
�� Ϊ���ɣ�(����HeΪ��������)
Ϊ���ɣ�(����HeΪ��������)
�� �˹��̵�ʼ��̬�ɱ�ʾ����
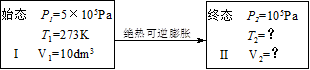
����������ʵ���Ϊ
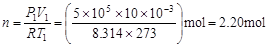
������Ϊ��ԭ�ӷ����������壬��

������Ϊ��ԭ�ӷ����������壬��
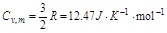
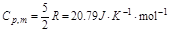
(1)��̬�¶�T2�ļ��㡣TV g�C1 = ���� = K PVg = ���� = K¢
P1�CgTg = ���� = K²
������T2�������֪���˹��̵�T��p��ϵʽ���������������
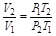
��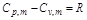 �����˶�ʽ����(1-69)ʽ���ɵ�
�����˶�ʽ����(1-69)ʽ���ɵ�
 (1-78)
(1-78)
����֪���ݴ�����ʽ

���� 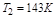
����̬�¶�Ϊ�C130��
(2) Q=0
(3)W�ļ���
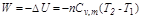
�� 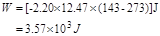
(4)�ļ���
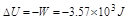
(5)�ļ���
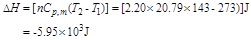
����1-11 �������Ĺ���Ϊ���Ȳ�������̣��ں㶨��ѹΪ105Pa�¿������͵�����ѹ��Ϊ105Pa���Լ���T2��Q��W�� ��
�� ��
��
�� �˹���Ϊ���Ȳ�������̣�ʼ��̬�ɱ�ʾ���£�

(1)T2�ļ���
��Ϊ�Ǿ��Ȳ�������̣���(1-69)��(1-70)����1-72����ʽ�������ã���Ӧ����(1-77)ʽ
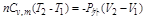
������������T2��V2����δ֪��������Ҫһ������T2��V2�ķ���ʽ���ܽ�T2�������������״̬���̿������Ҫ��
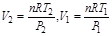
���˶�ʽ������ʽ�ɵ�
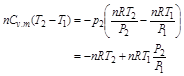
���ǵ�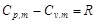 ��������ʽ�ɵ�
��������ʽ�ɵ�
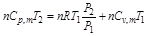
�� 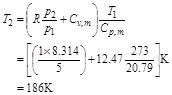
(2) Q=0
(3) W�ļ���
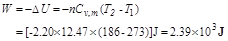
(4) �ļ���
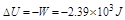
(5)�ļ���
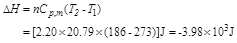
�Ƚϴ��������Ľ�����Կ�������ͬһʼ̬�������������ȿ�����̺;��Ȳ�������̣��ﲻ����ͬ����̬����������̬��ѹ����ͬʱ�����ڲ�������̵Ĺ�������Щ���ʲ����������̬���¶ȱȿ��������̬���¶ȱȿ��������̬���¶�Ҫ��һЩ��
2-11 ��������ѧ����֮��Ĺ�ϵ
��������Ҫ������S��U��H��F��G�������ѧ��������ȷ��Щ����ѧ����֮��Ĺ�ϵ�Լ������ʣ�Ѱ�ҳ���Щ����ѧ�������ض��������ÿ�ֱ�Ӳ�������ϵ��ѧ����ʾ�ĺ�����ʽ��������Ҫ�ļ�ֵ��
һ��������ʽ
��S��U��H��F��G֮��Ļ�����ϵʽ��
H = U + PV
F = U �C TS
G = H �C TS
��������ʽʵ�ʾ���H��F��G�Ķ���ʽ��
������ѧ��һ������
dU = dQ �C dW
���ڷ����ϵ�з�������������ı仯���̣�������ѧ�����ı仯�ɿ���;�����㣬�����
dW = �CP dV
ͬʱ������ѧ�ڶ�������
dQ = T dS
��
dU = T dS �C P dV ��2-48��
����H���ɶ���ʽ��
dH = dU + P dV + V dP= T dS + V dP ��2-49��
�Ժ�ķ����������F���ɶ���ʽ��
dF = dU �C T dS �C S dT= �CS dT �C PdV ��2-50��
�Լ���˹������G���ɶ���ʽ��
dG = dH �C T dS �C S dT = �CS dT + V dP ��2-51��
�����ĸ���ʽ���������������ѧ����֮��Ļ�����ϵʽ���dz���Ҫ���Ժ��һ�й�ʽ���������ĸ���ʽ�Ƶá�
�������ĸ��ֹ�ϵʽ�������Ȼ�ù������з������ı�ʾ��ʽ��������¶�T��
T = 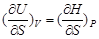 ��2-52��
��2-52��
������S��
S =  ��2-53��
��2-53��
����ѹ��P��
P = 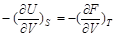 ��2-54��
��2-54��
�������V��
V = 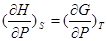 ��2-55��
��2-55��
�������˹Τ��ϵʽ
����ѧ��������ȫ�ֵ����ʣ��Ӷ�����U��H��F��G�ɸ����ֹ�ϵʽ�ó����¹�ϵʽ��
�躯�� z �Ķ�������Ϊx��y�� z����ȫ������
����
��������U�������ֹ�ϵʽdU = T dS �C P dV��
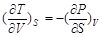 ��2-56��
��2-56��
������H�������ֹ�ϵʽ dH = T dS + V dP��
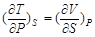 ��2-57��
��2-57��
���ں�ķ����������F�������ֹ�ϵʽdF= �CS dT �C P dV��
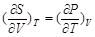 ��2-58��
��2-58��
���ڼ���˹������G�������ֹ�ϵʽdG = �C S dT + V dP��
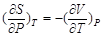 ��2-59��
��2-59��
������ʽ�ͳ�Ϊ���˹Τ��ϵʽ��
����Ӧ��
1����������ϵ����������ı仯
��dU = T dS �C P dV��
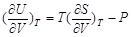
ͬʱ��dF = �CS dT �C P dV֪
�Ӷ�
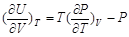 ��2-60��
��2-60��
����������ϵ��״̬���̣����������������ϵ����������ı仯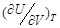 ��������������壬����״̬����PV=nRT����= 0������Ǹ�.�����ˡ�����ʵ��Ľ����
��������������壬����״̬����PV=nRT����= 0������Ǹ�.�����ˡ�����ʵ��Ľ����
2������������ѹ���ı仯
��dH = T dS + V dP��

ͬʱ��dG = �C S dT + V dP��
�Ӷ�
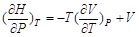 ��2-61��
��2-61��
5��������CV������仯�Ĺ�ϵ
�����������Ƶķ���������֤��
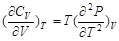
�Ӷ���
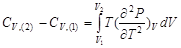 ��2-67��
��2-67��
���������۵Ļ����ϣ��ɽ�һ������CP�CCV�Ĺ�ϵ���ɵ�һ��֪��
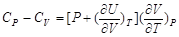
�����ϵʽ
����
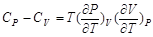 ��2-68��
��2-68��
P69��3-1 �Ҵ��ͼ״���ɵ���Һ�ɿ�������Һ�������20��ʱ�����Ҵ��ı�������ѹΪ5.93kPa�����״��ı�������ѹΪ11.83kPa��
(1) ����״����Ҵ���100g��ɵ���Һ�У��������ʵ�Ħ��������
(2) ����Һ��������ѹ�������ʵķ�ѹ��
(3) �״��������е�Ħ��������
�� 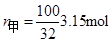
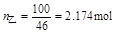

(1) 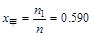
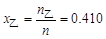
(2) 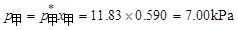

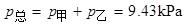
(3) ������

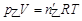
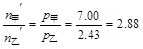
����
�ڶ�ƪ��������ѧ�߷ֿ����ܽ�ʼ�
������ѧ�ܽ�ʼǣ����������׳������ص��⣩
��һ�� ����ѧ��һ����
1�������ݵ����ٷ�ѹ����p����B pBѹ�����мӺ��ԣ�����ǹ������ʡ�����һ������ȷ��Ϊʲô�� �𣺲��ԡ�ѹ�����¶�һ����ǿ�����ʡ������мӺ��ԣ���ν�Ӻ��ԣ���ָһ������ѧƽ����ϵ�У�
ij���ʵ���������ϵ�����ʵ����������ȣ��� Cp����nBCp��m(B)���������ٷ�ѹ�����еķ�ѹpB ��ָ��һ���¶��£����B����ռ�л��������ͬ���ʱ�����е�ѹ������ѹ���ѹ�Ĺ�ϵ����ͬ һ����ѧƽ����ϵ������֮��Ĺ�ϵ�������ʵ������������ȹ�ϵ����p����pB�����Ӻ��ԡ����� ���������ǰѻ�������� ��ѹ p�����ַ�ѹ pB ��ϵ����Ϊ������ѧƽ����ϵ�������벿�ֵĹ�ϵ��
2����������ϵ���¶�����ʱ��һ�����ȣ����¶Ȳ���ʱ����ϵ�Ȳ�����Ҳ�����ȡ�������˵���Է�
��ʵ��˵����
�𣺲��ԡ����磺����������ѹ�����壬��ϵ�¶����ߣ�����δ�ӻ��������ȡ����磺�ھ���������
�У��� H2SO4 ע��ˮ�У���ϵ�¶����ߣ�����δ�ӻ������ȡ����磺��������������ͣ��ӻ��� �����ȣ���ϵ�¶Ȳ����仯�����¶Ȳ���ʱ����ϵ���Է��Ȼ����ȣ����ʱ��������������ˮ�� 1atm��100���±��ˮ�������¶Ȳ��������ȡ�
3����p(��)dV�룭p(��)��V�кβ�ͬ����pV���������������Ϊʲô����2�У�pVm(g)��������� �𣺣�p(��)dV ��ָ����С�����������p(��)��V ����ָ��ѹ����Ĺ����������������ѹ p ��
��Ĺ���������� V1�仯�� V2(��V��V2-V1)ʱ�����������pV������������������ָ����ѹ (p��)�����£���ѹp������仯ֵ��dV���ij˻���V�� dV �Dz�ͬ�ģ�ǰ����ָ��ϵ��������� ��������ı仯ֵ������仯ʱ�������������2�еģ�pVm(g)ʵΪ��p��Vm(g)-Vm(l)�ݣ���������� ��Vm(l)�������Vm(g)ʵΪ��V��Vm(g)-Vm(l)����ˣ�pVm���������
4���������������ܾ����������������ǵ�������ͬ����仰��ȷ��
�𣺲���ȷ����Ȼ�����������ܶ������������٣����������ϲ�ͬ����������ϵ�ı������ʣ���״
̬�����������빦����ϵ�뻷���佻������������������ѧ��������ϵ�Ĺ��������������DZ� �����������ݡ��е�������������ϵ���������ʣ�����״̬���������������ʲ�ͬ�����빦 Ҳ�����������������˶������ݵ������������������˶������ݵ�������
5. Ϊʲô���̲�������ѧ��һ���ɱ���ʽ�ǣ���U��Q��W������Щ���в��æ�U��Q-W�� �����Ƿ���ì�ܣ�Ϊʲô?
����Ϊ���̲Ĺ涨����ϵ����Ϊ��������Ϊ������ϵ����������WΪ��ֵ����������ϵ������
WΪ��ֵ���ܵ���˵����ϵ�ڹ����еõ�����Ϊ����ʧȥ����Ϊ����������涨�£�Ҫ�� �������غ�ԭ�������������ϵ�����ȼ��ϻ�������ϵ���Ĺ��ŵ�����ϵ���ܵı仯ֵ�� �����Ǧ�U��Q��W������Щ���ϣ����ķ����������涨�෴������ϵ��������W Ϊ��ֵ�� ��������ϵ������W Ϊ��ֵ��������Ǧ�U��Q��W��
6��һ��ϵ��A̬��B̬����;������� 100J������ϵ���� 50J����(1)��A̬��;����B̬��ϵ
���� 80J����QֵΪ���٣�(2) ����ϵ��B̬��;����ص�A̬��50J������ϵ���Ȼ��Ƿ��ȣ� QΪ���٣�
��(1) ��UA��B����100��50����50J Q����UA��B��W����50��(��80)��30J
(2) ��UB��A������UA��B��50J Q����UB��A��W��50��50��0
��ϵ������Ҳ����
7����֪��ϵ��״̬����ʽ��(T��p��V)��0����U��f(T��V)д����ѹ������ʱ��������ܶ��¶ȵ�
�仯�ʵı���ʽ��
��dU��(��U/��T)VdT��(��U/��V)TdV
ѹ������ʱ�����ԣ�T��(��U/��T)p��(��U/��T)V��(��U/��V)T(��V/��T)p
8��Ϊʲô��������ĵ�ѹ���̵��ȣ�ֻ��������ϵ�ij�����̬��
����Ϊ���������ĵ�ѹ������Qp����H���� ��H ����ϵ״̬�����ĸı�ֵ�����Сֻ��������
ϵ��ʼ��̬�����������������ĵ�ѹ���� Qp ��Сֻ�����ڳ���̬��
9������ ��H��Qp������ֻ�е�ѹ���̲��� ��H������仰�Ƿ���ȷ��
�𣺲���ȷ��H��״̬������H��U��pV��������ϵ״̬�����仯�����ܾ���ʲô���̣���ϵ����
ֵ�����ܱ仯���� ��H �п��ܲ������㡣
10����Ϊ����H��Qp������Qp Ҳ����״̬���������ʡ�����Ϊʲô��
�𣺲��ԣ���H��Qp��ֻ˵��Qp ����״̬����H�ı仯ֵ ��H��������ֵ����ȣ�������ζ��Qp
����״̬���������ʡ���H��Qp ֻ��˵�ں�ѹ����������������ض������£�Qp ����ֵ������ ϵ״̬���� H �ĸı䣬��������Ϊ Qp Ҳ��״̬������
11����֤������������ĵ��ݹ�������ϵ�Ħ�U��QV��
֤������U��Q��W ����ʱ ��V��0��������������W=0 �� ��U��QV
12��Ϊʲô�����������壬��ʽ��U=��nCV��mdT ������������һ���̵Ħ�U��
�����ܶ������������ƣ�
����Ϊ���������壬U��f(T)�����ܽ����¶ȵĺ�������ʼ̬���������۾�ʲô���̣�
�ﵽ��ͬ����̬��ֻҪʼ��̬�¶ȷֱ���ͬ����U ��һ����ͬ�����Թ�ʽ��U=��CV��mdT �����ܶ������������ơ�

���ݹ��̦�U1����CV��mdT ������̬���¶���ͬ
��ѹ���� ��U2����U1����U3 �� ��U3=0 �� ��U2����U1����CV��mdp �� 1mol�������岻��ʲô���̣�ֻҪ�䵽��ͬ�¶ȵ���̬�䦤U ���ǵ�����CV��mdT
13��Ϊʲô�������峣��R����ֵ�ϵ���1mol������������1Kʱ�����ĵ�ѹ������� ��W����p�⦤V����p(V2-V1)����nR(T2-T1)
�� n��1mol T2��T1��1K ʱ W��R
14����ϵ����100��N2����ȫת����NH3���簴��������ʽN2��3H2��2NH3������=?���簴����
����ʽ��N2����H2����NH3������=?���練Ӧǰ��ϵ��N2�����ʵ���n(N2)��10mol���ֱ� ��������������ʽ���õĦ��μ��㷴Ӧ��� n'(N2)����
��nN2(0)��100/28��3.57mol nN2(��)=0
����1��[nN2(��)-nN2(0)]/��B��(0-3.57)/(-1)��3.57mol
����2��(0-3.57)/(-1/2)��7.14mol
��ʽ��nB(��)��nB(0)+��B���� nB(0)��10mol
������ʽ��N2��3H2��2NH3�� nN2(3.57)��10-(-1)?3.57��6.43mol
������ʽ����N2����H2��NH3�� n'N2(7.14)��10-(-1/2)?7.14��6.43mol ���߽����ͬ��
15. ����Qp��m��QV��m+�Ʀ�B(g)RT��Qp��m һ������QV��m ��Ϊʲô������˵���� ��Qp��m ��һ������QV��m�����С�Ƚ�ȡ���� �Ʀ�B(g) �ķ��ţ����Ʀ�B(g)>0�� �� Qp��m�� QV��m������ �Ʀ�B(g)��0�� Qp��m��QV��m
���磺H2(g)����O2(g)����H2O(l)
��Hm��Qp����285.9 kJ?mol-1 �Ʀ�B(g)����1.5��0
QV��m��Qp��m���Ʀ�B(g)RT����285.8?103��1.5?8.314?298����282 kJ?mol-1 Qp��m��QV��m
�����磺Zn(s)��H2SO4(aq)����ZnSO4(aq)��H2(g)��
Qp��m����177.9 kJ?mol-1 �Ʀ�B(g)��1��0
QV��m��Qp��m���Ʀ�B(g)RT����177.9?10-3��8.314?298����180.37 KJ?mol-1 Qp��m��QV��m
16.���ȶ���ֵ����ֵ�����㡱����������Ħ�������Ⱦ��� 1mol �����������е���ֵ�� ����Ϊʲô��
�𣺲��ԡ��ȶ����ʵ���ֵ���������㡣������˵��״̬���ȶ����ʵĹ涨��ֵ�� ���㣬��Ϊ�涨��״̬�£��ȶ����ʵ������ʣ����涨��Ϊ0���������Ħ���� ���Ȳ��� 1mol ���������е��ʵľ���ֵ��������������������ȶ����ʵ��ʵ��� ��ֵ�������Ա�״̬���ȶ�����������Ϊ�������ߣ��ó������ֵ��
17. ֤���ɼ��ʼ��㷴Ӧ�� ��Hm �Ĺ�ʽ�ǣ���rHm��(-��ni��i)(��Ӧ�����)
�𣺻�����Ħ�fH������ni(��H��ԭ��)��(��nj��j)
����Ӧ��ЧӦ ��rHm���Ʀ�B(��Hm��f��)B���Ʀ�B[��ni(��H��ԭ��)����(nj��j)]B ���Ʀ�B(��ni��H��ԭ��)B���Ʀ�B(��nj��j)B
����ɲ����뷴Ӧ���Ԫ����ͬ���Ҹ���ԭ�ӵ���ĿҲ��ȣ�
�� �Ʀ�B(��ni��H��ԭ��)B��0
���Ц�Hm�����Ʀ�B(��nj��j)B
�����Ʀ�B(��nj��j)B(��Ӧ��)���Ʀ�B(��nj��j)(����)
������Ӧ��ļ���ϵ����B����Ϊ��ֵ������ʽ(-�Ʀ�B(��nj��j)B(��Ӧ��)�����Ϊ �Ʀ�B(��nj��j)B(��Ӧ��)���ٽ�һ��B�����еģ����nj���Ϸ���ʽ�з�Ӧ��ļ� ��ϵ���ͣ����Ǹ÷�Ӧ�����з�Ӧ���ܣ����nj����дΪni������ ��i����ô��
��Ӧ����ܼ���ֵ��Ϊ(��ni��i)(��Ӧ��)��ͬ���Բ���ļ���ϵ������Ϊ��ֵ��
��Ϊ (��ni��i)(����)����ã���Hm=(��ni��i)(��Ӧ��)��(��ni��i)�������
18. ��Ӧ A(g)+2B(g)����C(g) �Ħ�rHm(298.2K)��0����˷�Ӧ����ʱ�ض����ȣ�
���� Ϊʲô��
�𣺲��ԡ�ֻ���ڵ�ѹ�£��������ʱ��Qp����Hm����Hm��0���� Qp��0����ϵ�ض� ���ȡ������з�����������߷ǵ�ѹ�����£���Hm��Qp ����Hm��0��Qp����С��0�� ����0����һ�����ȡ����磬����������H2���2 ȼ�գ���Hm��0����Q��0�� �����ȡ�
19. ���������һ����ѭ�����̣�ѭ������һ���ǿ�����̡�����˵������? Ϊʲô? �𣺲��ԡ�������̲�һ��Ϊѭ�����̡���ΪֻҪ��ϵ��A̬����Ħ��������ЧӦ����
������£�����һϵ�����ӽ�ƽ��״̬����B̬������A��B�Ĺ����ǿ��档��Ȼ�� �����̬ A ����̬ B ��������ͬ��״̬����A��B�㲻��ѭ�����̣����B̬����
A ̬��ù��̱��ǿ���ѭ�����̡�ѭ�����̲�һ���ǿ���ģ���ʼ̬A��ʼ��״̬�� ���仯������;���������ֻҪ�ص�ʼ̬A������ѭ�����̡�ֻ�ǣ���A̬��ʼ�� ����Ħ��������ЧӦ���ڵ�����£�������һϵ�����ӽ�ƽ��״̬���ֻص�A̬�� ѭ�����̲��ǿ���ѭ�����̡���֮���������ѭ��������������ȫ��ͬ�ĸ��
20. ����ͬһ��̬(p1��V1)�����ֱ��¿���ѹ������ȿ���ѹ��������̬����̬�� ������V2����һ����������ѹ������Щ��Ϊʲô��
��(�涨��������Ϊ��ֵ)�����ȿ���ѹ�������ڵ��¿���ѹ������������Ϊ����ѹ�� ʱ������������ȫ���������������ܣ����������¶����ߣ��ʵ�������̬���Ϊ V2 ʱ�������ѹ���Ⱦ����¿��浽��V2 ʱ�����ѹ��Ҫ�ߣ������ȿ���ѹ��ʱ�� ����ʩ�ӵ�ѹ����Щ���������ѹ����Ҳ��Щ��
21. ��ͬһ��̬(p1��V1)�ֱ���ľ��������벻����ľ�����������̬�������V2 ʱ������ѹ����ͬ��Ϊʲô��
�𣺲���ͬ��������������ɣ�p1��V1����V2 ��ϵ�����Ĺ����ڲ�������������� ��p1��V1����V2 �������Ĺ����������̵� Q �������㣬���ǰһ��������ϵ����
���͵ø��࣬��Ӧ��̬������¶�Ҳ��Щ�����Կ���������ͱȲ�����������͵� ��̬V2 ʱ�����ѹ����Щ��
22. �������徭һ����ѭ�����ܷ�������ת��Ϊ��������ǵ��¿���ѭ���������� �𣺲��ܡ���������������ڵ��¹����в��䡣��U��0
����ѹ����������
�������� A(p1��V1��T1)��������������������B(p2��V2��T1)
������ W(��)����Q(��)����p2(V2��V1)���پ�������ѹ���ص�ʼ̬��
����ѹ��
B(p2��V2��T1)�������������� A(p1��V1��T1)(ԭ��̬)
W������Q������RTln(V1/V2) ����Ϊ����ѹ���������ĵĹ���С��
����ѭ�����̣�
W��W(��)��W������p2(V2-V1)��RTln(V1/V2)����Q
�� ��p2(V2-V1)��0����RTln(V1/V2)��0������ǰ�ߵľ���ֵС�ں��ߣ�
��W����Q��0��Q��0���������ȣ�W��0��ϵ�ù���������ʧ�ȡ�
˵������ѭ�������У���������ϵ���������õ��ǵ������ȣ����ǰѻ������ȱ�ɹ��� ͬ�������A����B�ǵ��¿������ͣ�B����A�ǵ��²�����ѹ�������Ҳ��W��0�� Q��0����ϵ�ù����������ȣ��������������õ��ȡ����ܰѻ����ȱ�ɹ���
��� A����B�ǵ��¿������ͣ�B����A�ǵ��¿���ѹ������Ϊ���¿���ѭ�����̣� W����RTln(V2/V1)��RTln(V1/V2)��0�� �� Q����W��0����������ϵ���ǻ�����
��δ��ʧ��������״̬δ�䡣
���Ϸ������������徭һ����ѭ�������ܽ�������ת��Ϊ����
23. ���������ڵ�һ���������Ŵ�ʹ���������ת���ܷ�ȫ���¶ȣ���û��� 0�� ������(25��)���������ѭ��ÿСʱ�ܶ��� 1Kg �ı����緿���������Ϊ 150 KJ?K-1���������乤�� 10 Сʱ�������¶ȵı仯��
�𣺲��ܡ���Ϊ�������Ŵ����������ڿ�����ͨ��ʹ�ߵ�������Դ�¶���ȡ������
��������������ĵĵ繦�Լ���������ȴ�� (������Դ)�����ȶ����ȵ���ʽ�ŵ��� �� (������Դ)���������Ŵ�ʱ�����ڿ������������ڣ�ʹ�����������ߡ������� �ܵ�Ч������������ĵ���ת��Ϊ���ڿ��������ܣ���ʹ�����¶����ߡ����ʹ�� ���¶ȷǵ������ͷ������ߡ�
1��ˮ�ı���Ϊ 4.184 J?K-1��1��ˮ��������Ϊ 339 J?g-1
Q'��1000?(4.184?25+339)?10��4436 KJ
�£�T1��(T2-T1)= 273/25 = 10.92 �£�Q'/W
W��Q'/10.92��4436/10.92��406.23 KJ
Q2��W-Q'��4842 KJ ��T��4842/150��32 K
T3��298+32�� 330 K ,�����¶ȱ�Ϊ330K��
�ڶ��� ����ѧ�ڶ�����
1. ʲô���Է����̣�ʵ�ʹ���һ�����Է����̣�
����ϵ����Ҫ����������������Ϳ��ܷ����Ĺ��̽��Է��Թ��̣�������ϵ������ �ϻ�ʵ���������������������Ĺ��̽��Է����̡�ʵ�ʹ��̲�һ�����Է��Թ��̣� ����ˮ���Dz������Է��ԵĹ��̡�
2. Ϊʲô����ѧ�ڶ�����Ҳ�ɱ���Ϊ����һ��ʵ�ʹ��̶�������ѧ������ġ���
������ѧ�ڶ����ɵľ����������ʵ�����漰�������빦ת����ʵ�ʹ��̵IJ������ԡ� ��ʹ���̵IJ������Զ����������������ȵ�ת�������ǿ���ģ���ô���е�ʵ �ʹ��̷����������ºۼ�����Ҳ��Ϊ������ˣ��������Ʒ�������ѧ�ڶ����ɣ� Ҳ�����ȹ�ת���IJ������ԣ���ʵ�ʹ��̶��Dz�����ġ�Ҳ��������������á� һ��ʵ�ʹ��̶��Dz�����ġ�����������ѧ�ڶ����ɡ�
3. ������̵����������ر��Ƿ���ȣ�Ϊʲô? ���ɹ��̵����������ر��Ƿ���ȣ� �𣺿�����̵������̼������ر䡣����S��QR/T (��S���Ҧ�QR/T)��������������� �����ر䲻�ȣ���ԭ�����ڿ�����̵� QR ���� QIr������ʵ���Dz���������ر� ����������Դ��һ���������̣���һ������Ħ���Ȳ�����������ɵġ���ˣ������� �����ر���������̡���������״̬�������ر䲻�۹��̿������һ��ʼ��̬ȷ��,
��S ֵ��һ���ġ�
4. Ϊʲô˵(2-11)ʽ�ǹ��̷���Ĺ�ͬ�о�? Ϊʲô˵��Ҳ�ǹ��̲�����̶ȵ��о�? ��(2-11)ʽΪ����SA��B����A����Q/T��0������ʵ�ʹ����Dz�����ģ���ʽָ����ʵ
�ʹ���ֻ���� ��SA��B����A����Q/T ������ķ�����У��� ��SA��B����AB��Q/T С���� �Ĺ����Dz����ܷ����ġ����(2-11)ʽ����Ϊ���̷���Ĺ�ͬ�оݡ��������Է����̷� ����оݣ�(��S-�Ʀ�Q/T) �IJ�ֵԽ����ʵ�ʹ��̵IJ�����̶�Խ��������Dz����� �̶ȵ��оݡ�
5. ������Щ˵���Ĵ�������� Ϊʲô����������Ĵ���д����ȷ��˵���� ��B
(1)��Ϊ��S���� ��QR/T������ֻ�п�����̲����ر䣻����S���Ʀ�QIr/T�����Բ��� ��A
�����ֻ�������̣�����û���ر䡣
(2) ��Ϊ��S���Ʀ�QIr/T��������ϵ�ɳ�̬ A ����ͬ�IJ�������̵�����̬ B������ �ı�ֵ������ͬ��
��B
(3) ��Ϊ��S������QR/T������ֻҪ������̬һ�������̵������̵�ֵ����һ���ģ� ��A
��� ��S ��һ���ġ�
��(1) ����״̬��������S��SB��SA ����ϵ�� A ̬�� B ̬��仯ֵ ��S��һ���ģ��� ���̵Ŀ�������أ����������ǹ���������A̬��B̬���̵IJ�����̶Ȳ�ͬ���� ��������ֵҲ����ͬ���������������ԭ�����ڶ��ص�״̬�������ʲ����⣬���ر��� ��B
���������������ʲ�ͬ�ĸ����Ϊһ̸����S���� ��QR/T��ֻ˵������������ֵ����
��A
�ȣ������Ǹ����ϵ�ͬ��
(2) ��Ϊ����״̬�������۹��̿�������䦤S��SB��SA��ֻҪʼ��̬һ������ֵһ���� ��ı�ֵ������ء�����ԭ������û���պ�״̬�����ĸ��
(3) �������ڽ���������������״̬�����ı�����Ϊһ̸��ʼ��̬һ���������̿����� ������ֵ����ȷ��˵���ǣ�ֻҪʼ����̬һ�����䦤S �ı�ֵ��һ���������̵�ȴ�� ���̵IJ�����̶Ȳ�ͬ����ͬ�������п�����̵����������������ر䦤S��
6.�����ھ��ȹ����Ц�S��0����ĩ��A̬�������������벻������̶�����B̬������ͬ һ״̬B����������ͬ����ֵ���ؾͲ���״̬�����ˡ�����Ȼ����һ�����Ǵ���ģ� ���ںδ�����������������������̲���֮��
�𣺾��ȿ�������Ц���ֵһ�������㣬��˸ù�����QR��0����ϵ�뻷�����Ƚ����� �����Ȳ���������У�QIr��0��������һ�������㣮���⣬��ͬһʼ̬���������� �����������Ȳ�������̴ﵽ����̬�Dz�ͬ���������������ͬһʼ̬�������ֱ� �������ȿ������ͺ;��Ȳ��������ʹﵽ��ͬ��ѹ�������ȿ����������������Ĺ� �ľ���ֵ�Ⱦ��Ȳ�������������������Ĺ��ľ���ֵҪ��Щ�����ܽ��͵�Ҳ��Щ���� ���ȿ��������̬�¶ȵ��ھ��Ȳ����������̬�¶ȣ���ͬ����̬ѹ��ʱ����̬��� �Ǿ����ȿ�����̵�С�������Ȳ�������̵Ĵ������Dz�ͬ����̬��
7. 263K �Ĺ���ˮ��� 263K �ı�����S��0����������ԭ����ì����Ϊʲô��
�𣺲���ì�ܣ�������ԭ�����������ǹ�����ϵ�������ϵ�����������̲����߱������ ����������ϵ�뻷�������Ƚ��������ǹ�����ϵ�������ϵ����S ����С���㡣���� �ػ������ġ�
8.��p����298K�����ˮ������� 298K ��ˮ���ŵ���Qp��Qp����H������Hֻ������ ������̬�����ѹ���̵Ŀ�������أ��������ø������̵��� Qp������ ��S�� Qp/T (TΪ298 K)��������ϵ���ر䡱���ֿ����Ƿ���ȷ��Ϊʲô��
�𣺲���ȷ������ֻ�ܵ��ڿ�����̵�������֮�ͣ�����˵����ͨ��������̵������� �������ر䦤�ӣ�����������Ϊ��������¹��̣��ʦ��ӡ�Qp/T���������������� ������ϵ�Ħ��ӡ�
9. ����һ��ѧ��Ӧ���ѹ��ЧӦ��H��0����÷�Ӧ����ʱһ�����ȣ��Ҧ�S��0������ Ϊʲô��
�𣺲��ԡ���Ϊ��ѧ��Ӧ����ЧӦ��H��ָ�ڵ��µ�ѹ��������������£���ʱQp�� ��H������H��0��Qp��0����Ӧ����ʱ���ȡ������Ӧ�����ڵ��µ�ѹ������� ���������£�Q�٦�H����H��0��Ҳ��һ�����ȡ����磺����������H2���2ȼ�� ��Ӧ����Ӧ�ĵ�ѹ��ЧӦ��H��0������������Q��0�������ȣ�Ҳ�����ȡ������ �µ�ѹ���ڿ����ط����ķ�Ӧ����Ȼ��H��0����Q���ܴ����㡣��ʹ�Ƿ��ȷ�Ӧ�� ����Ҳ��һ��С���㣬���磺Ũ H2SO4 ����ˮ�����ȣ��� ���ӣ�0��
10. ����S��ln�������������ڿռ��������ֲ��ϻ��ҳ̶ȵ����ȣ����ж������� �µ�ѹ���̵Ħ�S�Ǵ�����? С����? ���ǵ�����?
(1) NH4NO3(s)����ˮ�� �𣺦�S>0
���� (2) Ag(aq)+2NH3(g)����Ag(NH3)2�� �𣺦�S<0
(3)2KClO3(s)����KCl(s)+3O2(g)�� �𣺦�S>0
(4)Zn(s)+H2SO4(aq)����ZnSO4(aq)+H2(g) �𣺦�S>0
11. ���ʵı��أ���(298K)ֵ���Ǹ�״̬���صľ���ֵ��
�𣺲��ԡ����ʵı��� S����298���Ծ������ 0K ʱ�����������ֵ�涨Ϊ����Ϊ
���㣬������ڱ�ѹ��p���� 298K �� 0 K ����ֵ֮���ˣ�S��(298K)��ָ�� ѹ�� p���¡�298K ����ֵ����� 0K ʱ��ֵ�����ֵ�����Ǿ���ֵ��
12. (2-29)ʽ��(2-32)ʽ���������кβ�ͬ? Ϊʲô��(2-32)ʽ�ж����̵��Է���ʱ���� �������������������?
��(2-29)ʽ�� dGT��p��W'=0��0��(2-32)ʽ����GT��p��0��(2-29)ʽ�ǵ��µ�ѹ��
��������̵��Է������оݣ����������ڸ�������ʵ�ʹ�������ϵ����˹�����ܽ��� ������У�����ϵ�������ܲ��ٸı�ʱ��ﵽƽ��̬��������˹��������������Dz� ���ܷ����ġ�(2-32)ʽ�ġ�������ʾ�Է��ԣ�����������ʾƽ��̬���ڵ��µ�ѹ�²� ����ϵ�Ƿ�������������Է����������ؼ���˹�����ܽ��ͷ�����У�ֱ�� G ֵ���� �ﵽƽ��̬����� W'��0�������� W' ������ ��GT��p��0��������������ϵ�� W'
����W������ֵС�ڦ�G����ֵ���������Է�����ʱ����GT��p��0����� W'��0�� ��GT��p��0�Ĺ��̲��ܷ�������ϵֻ�ܷ����Է����� ��G �� 0���ɴ˿ɼ��������� ϵ�Ƿ������������(2-32)ʽ���ǵ��µ�ѹ���Է����̷������ȵ��оݡ�
13. ����GT��p��W'=0��0 ˵����G<0 �Ĺ���ֻ����T��pһ������W'=0 �������²��ܷ������� ����˵������? Ϊʲô?
�𣺲��ԡ���GT��p��W'=0��0��˵����T��pһ��ʱ����������������¦�G��0�Ĺ��� �����Է����У����ù��̲���ֻ���� W'��0�����·������з������ W' ʱ��ֻҪ ������������ľ���ֵС�ڼ���˹�����ܵĽ���ֵ������Ҳ�ܷ�����
14. ���ڹ�ʽ��GT��p��WR'������˵���Ƿ���ȷ��Ϊʲô��
(1) ����ϵ�� A ̬�� B ̬���۽���ʲô���� ��G ֵΪ��ֵ��һ������W������
(2) �����µ�ѹ��ֻ����ϵ�������������ʱ G �Ž��͡���
(3) ��G������ϵ�����������������һ������������
��(1)���ԣ�ֻ���� T��p һ���Ŀ�������У���ϵ�� ��GT��p�ŵ��� WR������������ ���£������������ ��GT��p ������ WR����
(2) ���ԣ���ϵ�ļ���˹��������״̬����������GB��GA������A��B�ĵ��µ�ѹ�� �����Է��ģ���������ʵ�ʹ�������ϵ�Ƿ�����������������ϵ�����ܶ��ǽ��͵ġ�
(3) ���ԣ�ֻ���ڵ��µ�ѹ�����£�����˹�����ܵĽ���ֵ�����������������ֵ��
15. Ϊʲô���µ�ѹ�»�ѧ��Ӧ���Է��Բ����æ�H���оݣ�����Щ������æ�H���оݣ� ���ܵõ���ȷ�Ľ��ۣ�
�𣺵��µ�ѹ�»�ѧ��Ӧ�Է��Ե��о����æ�G���������æ�H���������Ц�G����H�� T���ӵĹ�ϵ����˶ԣ���H������T��S���ķ�Ӧ���æ�H���о����õ��Ľ������� ��G �о���һ�µģ������������¿����� ��H ��Ϊ�оݣ����⣬���ڦ�H��0����S�� 0��H ��0�����ӣ�0�ķ�Ӧ���æ�H�о��� ��G �о�Ҳ��һ�µģ����Ҳ���� ��H����Ϊ�оݡ�
16. ���ڦ�H��0�����ӣ�0���ڳ����²����Է����еķ�Ӧ�ı��¶��ܷ�ʹ��Ӧ�Է��� �У�Ϊʲô��
���ܹ������ݦ�G����H��T���ӹ�ʽ��T��S ���¶ȵ����߶�������߷�Ӧ�¶�ʹ T��S����H���Ӷ� ��G��0����Ӧ�����Է������ˡ�
17. һ�����ֽ��������ʱ�������������ȣ��ԱȽ���������ڵ���������µ��ȶ��ԡ� �𣺹���ֽ��������ķ�Ӧ���ȣ���H��0�����ڲ������壬��S��0�������¶����ߣ� T��S ���ӣ���G����H��T���ӣ����¶����ߣ���Ӧ�� ��G ���ͣ����Ը����¹��� �����¶��Խϲ
18. Ϊʲô��U =��TdS-��pdV �����ڵ���־�������ϵ���κι���? ���Ƿ���ζ�Ŷ� ���ּ�����ѧ��ϵ���κι��̡�TdS����pdV���ֱ�������빦��?
�𣺶�dU��TdS��pdV��ʽ����һ��������־�����ϵ��ֻҪ������̬��ͬ�����۹��̿� ����������ã�����Ϊ����־�����ϵ���ᷢ����ѧ�仯����仯��ֻ��p��V��T �ı仯��ͬʱ������ʽ�У�U���ӡ�V��״̬��������仯ֵ������أ���˸�ʽ �������κι��̣�����ֻ���ڿ�������С�TdS ������ϵ�������ȡ�����pdV������ ϵ�������������
19. ���� ��Q��dU��pdV �� dU��(��U/��V)TdV��(��U/��T)VdT ��ȫ���б�ʽ֤��Q ����״̬������
��ȫ�ֵ�һ����Ҫ�����Ƕ����������Ĵ����أ���������ϵʽ����
dU �����Q������ (��U/��T)V��T(��p/��T)V��p ���룬
��Q��(��U/��T)VdT��[(��U/��V)T��p]dV �У� ��ô��
[��(��U/��T)V/��V]T��[��(��U/��V)T/��T]V��(��p/��T)V
���� Q ������ȫ�����ʡ�
20. �ֱ����۶�ѹ�������¶ȼ�����������ѹ��ʱ���¹��̵Ħ�Gֵ��α仯��
(1) �е���Һ������Ϊ������
(2) ���̵���Һ����Ϊ���壨��Vm(l)��Vm(s)����
������(����G/��T)p������S���ɦ���ֵ�����ж��ڶ�ѹ�¦�G ��T�ı仯�ʵġ� �� ��S��0 ʱ����(����G/��T)p��0�����¶����ߣ���G ֵ��С���� ��S��0ʱ����
(����G/��T)p��0�����¶����ߣ���Gֵ��������(����G/��p)T����V���ɦ�V ֵ
���ж��ڶ����£���G ��ѹ��p�ı仯�ʡ�����V>0 ʱ���� (������/��p)T��0�� �����£���ѹ������G���ӡ�
��1���ڷе���Һ������Ϊ��������S��0����ѹ�������¶����� ��G��С�������� ���У�����V��0������������ѹ������G�������������С�
��2��Һ�����̳ɹ��壬���� Vm(l) > Vm(s)����V < 0�������¼�ѹ���̹��� ��G ���ͣ����̸����С������ӣ�0����ѹ�����£���G�������̲����С�
21. ����� 1000cm3 ˮ�м���1mol H2SO4 ������Һ�����ΪV�������Һ�� H2SO4 �� ƫĦ�������V��1000cm3������
�𣺲��ԡ���Һ���Ϊ�� V��(1000/18)VH2O��VH2SO4���� H2SO4 ��ƫĦ�����Ϊ VH2SO4��V��(1000/18)VH2O������Һ��ˮ��ƫĦ����� VH2O��18 cm3��ˮ���� ��Ҳ������ 1000 cm3����VH2SO4��V��1000��
22. ��f��p�� ��״̬������ʵ����ı�̬������Ϊʲô��
�𣺲��ԡ���ʵ����ı���ָ�� f��p����101325pa�������������ַ�������������Ϊ ��״̬���� f��p�����ã�1�ļ���̬������ʵ����棽p��(��101325pa)��״̬����
��ϵ�� �� ��1����˲��DZ�̬��
. ��� 1000cm3 ˮ�м���1mol �� H2SO4����Һ��������Ӧ�V���� H2SO4��ƫĦ������ ֵ���� ��V��Ϊʲô��
�𣺲��ǣ����еĦ�Vֵ����Һ�仯����������� H2SO4 ��ƫĦ���������ΪƫĦ����� �Ķ����� V(H2SO4)=(��V/��n)T��p��n1����������Ϊ�������� H2SO4 ˮ��Һ�У����� 1mol H2SO4 ��������Һ����ı仯ֵ��V����������������������ϵ����˦�V ���� H2SO4 ��ƫĦ�������
2. ����ҺŨ������ʱ�����ʵ����ܽ�����Ħ�������ܽ��ȸ������ֵ����Ũ���� ������ϡ��ʱ���ֽ�������
�𣺵�Ũ������ʱ�����ʵ����ܽ����������㣬��Ħ�������ܽ���������ijһ��ֵ�� ��Ũ����������ϡ��ʱ�����ܽ��ȵ������ʵĻ����ܽ���������ԭ��ʱ��б�ʡ��� �����ܽ����������㣬��������Ħ�������ܽ�������һ����ֵ��
3. (3-6) ʽ�� (3-7)ʽ�������������������壿
��(3-6)ʽΪ����Hsol��Hslu��[nBH*B��(1/MA)?H*A]���������������ϵ�Ļ����� ���ȵ������Ϻ�(��̬)����ǰ(��̬)���ʲ�ֵ��
(3-7)ʽΪ��(����Hsol/��nB)T��p��n(B)��HB��HB��m ��������B�����ܽ��ȵ���B ����Һ�е�ƫĦ�������䴿̬��Ħ����֮�
4. ���ڶ���������������к���ͬ��
����ͬ�㣬���������������Һ����ֵ�����ѹ������������Һ�е�Ũ�ȣ�������ߵ� ��ѧ��ʽ��ͬ����ͬ�㣬���ڶ���������������ϡ��Һ���ܼ�����ʽ�еı�������Ϊ ���ܼ� A �������¶�ʱ�Ĵ� A ������ѹ p*A��������������������ϡ��Һ�е����ʣ� �������ɵı�������Ϊʵ��ֵ�ľ��鳣���������У�
Kx��lim pB/XB�� Km��lim pB/mB�� KC��lim pB/CB
������ x0 m0 c0
��������ֻ����ѧ�ϵļ��������������������塣ʹ�ú�������ʱ����Ũ�ȵ�λ�ɰ� ��ҪӦ�ã���m��c�����Һ������ɵ���ʽҲ����Ҫ��д��pB��RxXB��xB��KxpB �� ��ʽ��ʹ�õ��ܶȵ�λ��ͬ����������Ҳ��ͬ��
5. ����������ÿһ�������Ƿ���һ����ֵ��������Ũ�ȡ��¶ȡ��ܼ����ʶ�����
�𣺺���������ÿһ�����ʲ���һ����ֵ���Բ�ͬ�����ʣ��ڲ�ͬ���¶����ܼ��в�ͬ�� ��������ֵ����ͬһ�¶���ͬһ�ܼ����������������Ũ�ȵı仯���仯�������¶� �ı仯���仯�����⣬����������ʹ�õ�Ũ�ȵ�λ��ͬ����ֵ��ͬ������ͬŨ�ȵ�λ �ĺ�������֮����ת�����㡣
6. ��ν�ܼ�����ν���ʣ����߸����������Ĺ����ԣ�
���ܼ������ʵĻ��־�����Ե����塣����Һ������Һ���γɵ���Һ�������������� ���ܼ��������ٵ���ֽ����ʡ��������塢��������Һ���γɵ���Һ�����塢����� ���ʣ�Һ����ܼ���������ϡ��Һ���ܼ��������ڶ����ɣ����ʷ��Ӻ������ɣ����� ������У����ڶ����������������һ���£���������κ�Ũ���¶��������ڶ����ɡ�
7. Kx��Km��KC ����ʵ��ֵ�Ƿ����ϵ��������Ϊ�����ڵĹ�ϵΪ��
Kx��Km��MA��K��A��MA�������ŷ�
������ ��B��mB��cB ����Ũ�ȵ�λ֮�������ϵ����Kx��Km��KC ֮���Ȼ����
��ϵ����ϡ��Һ�������£�����Ũ�ȵ�λ�ĺ�������֮��������и����Ĺ�ϵʽ��
8. ��̬�������������壬�ܼ������ʵı�̬�к���ͬ���������Ҳѡ��Ħ��������B ΪŨ�ȵ�λ����ô���ı�̬������һ��״̬��(��ʾ���̲���ͼ��--����Kx��)
�����ʵı�̬һ���Ǽ���������ڻ�����У�������Һ�еĻ�ѧ�����ֵʱ��ѡȡ�Ļ�̬��
Һ̬������������һ������ B �ı�̬Ϊ��B��=��B*(l��T��p��)��ϡ��Һ���ܼ�A�� ��̬Ϊ����A(T)=��*A(l��T��p��)������B�ı�̬Ϊ��������ֱ�ߵ��ӳ�����ijһ״̬�� ��Ũ��Ϊ1��Ũ�ȵ�λ(xB��1��mB��1��cB��1�������õ�Ũ�ȵ�λ����)��������ϵ�� ��B(��x����m���C)��Ϊ1��һ������̬������ԣ�B��ʾŨ�ȣ���̬�ǣ�B��1�������Է��� �������ɵļ���̬����ͼ�������е� Kx�㡣
9. ��B(l��T��p)����B*(l��T��p)����B��(l��T)����B��(g��T) �ĸ����ŵ������кβ�ͬ�� �𣺦�B(l��T��p) Ϊ���¶�T��ѹ��pʱҺ̬���������� B �Ļ�ѧ�ơ�
��*B(l��T��p) Ϊ���¶�T��ѹ��pʱҺ̬�� B �Ļ�ѧ�ƣ�
����B(l��T) Ϊ�¶�T����ѹ��p���£�Һ̬������У����B�ı�̬��ѧ�ƣ� ����B(g��T) Ϊ�¶�T����ѹ��p���£���Ϊ��̬���������� B �ı���ѧ�ơ�
10. �������ߺ�����ȷ(1)����B��m(l)������B��C(l)�� (2)����B��m������B��C��RTlnKC��
(3)����B��C(l)������B��m(l)��RTln(KB��C/kB��m)��
�𣺵���ʽ��ȷ����ʽָ��������ϡ��Һ����� B ���ֲ�ͬŨ�ȵ�λ m �� c �ı�
�Ļ�ѧ��֮�
11. ���ݹ�ʽ (3-25)�� (3-26)������� ��B��m���B��C������1ʱ��״̬���DZ�̬��
����뷨�Ƿ�ԣ�����ͼ��������ͨ���Խ��� m=1 �ĵ���һˮƽ����ʵ���ཻ�ĵ㣬 �仯ѧ��Ϊ���٣��Ƿ��DZ�̬��
��(3-25)ʽ����B(l��T)������B��m(l��T)��RTln��B��m ��
(3-26)ʽ����B(l.T)=��B��C(l��T)��RTlnaB��C ����B��m���B��C ����1�ĵ㣬��״̬
��һ���DZ�̬��ֻ�л��ϵ��Ҳ�� �ã�1���DZ�̬��
ͼ�������У�ͨ���Խ��� m��1�ĵ���һˮƽ����ʵ���ཻ�ĵ��仯ѧ�ƵĴ�С��� ̬��ѧ����ȣ������DZ�̬��
12. Ϊʲôϡ��Һ�ķе����ߡ������½�����ѹ�Լ��ܼ�����ѹ�½���Ϊ�����ԡ����� �����Ե������ԭ����ʲô��
�������������ʵ���ֵֻ�������ܼ������ʵ������������йأ��������ʱ����������� �أ��ʳ�Ϊ�����ԡ������Բ�������Ҫԭ����������������������ܼ���ѧ�ƽ��͡�
13. ���ܼ���һ���������ʾ���ʹ��Һ������ѹ���ͣ��е����ߣ����㽵�Ͳ��Ҿ����� ѹ����仰�Ƿ�ȷ��Ϊʲô��
�𣺲�һ��ȷ���������������ǻӷ��Եģ����һӷ��Ա��ܼ�������Һ������ѹ�� �ӣ��е��½�����������Ƿǻӷ��Եģ��������ʵĻӷ���С���ܼ�������������� ȷ��
14. �����ˮ�м����������Ҵ������ĸ������Խ����������ı仯��Ϊʲô�������ı仯��
����� NaCl��CaCl2 ����������
��ˮ�м����Ҵ�����Һ����ѹ�������е��½������������½�����ѹ�Դ��ڡ�
���������Ҵ��ǻӷ��Եģ����һӷ����ֺܴ��Ҵ�ˮ��Һ����ʱ�������Ĺ�̬�� �Ǵ������������ NaCl������ÿ��������ȫ����ɶ������ӣ�������ѹ���ͣ��е� ���ߣ�������������ֵ�ӱ��������� CaCl2�����������������
15. ijһ��Ũ�ȵ�ϡ��Һ�������ĸ������Լ�����������ļ�����ϵ��
�𣺦�Tb/Kb����Tf/Kf����VA/(RTMA)��(mpA*/pA)/MA��
16. ���ù�ʽ(������0)ʱ������Һ�������Ĺ����Ƿ�����ǹ�̬���ܼ���Ϊʲô��
��(3-40)ʽΪ����Tf��KfmB�������DZ����̬���ܼ������Ƶ���ʽ�Ļ�������Һ��
�ܼ� A �Ļ�ѧ�����̬���ܼ��Ļ�ѧ����ȡ�
17. ����������������ֻ�����������е�ÿһ����ֲ�����ȫ��ɷ�Χ�ڷ�����
�ڶ����ɡ�
����Ϊ����������ÿһ���������������Χ�����������֮�������ã���ͬ�� ����ִ��ڴ�̬�����(������ͬ������֮��������)���������������ȫ�� �ɷ�Χ�ڷ������ڶ����ɡ�
18. ��Һ�е�����������е�����к�����?
����Һ���������塢��������Һ���γɵģ����塢�����Ϊ���ʣ�Һ���Ϊ�ܼ������� �����б���������Һ���϶��õġ�������ѧ����ʱ�����������һ��ֻ�ѧ�ƹ�ʽ �����ӻ���Ʒ������ڶ����ɣ�����Һ�е��ܼ������ڶ����ɴ��������ʰ��������� ����������������ò�ͬ�Ĵ������̡�
19. ��������������Һ̬(���̬)�����������Ӽ��������к�����
����������ķ��Ӽ䲻��������ã���������������Ӽ��������ã�������ͬ �����ӻ�����������֮���������һ����
20.������ϡ��Һ��������������һ���������仰����Ϊ��Σ�
������ϡ��Һ���������Ƶط��Ӻ������ɣ��ܼ��������ڶ����ɣ����ߵı�̬���в� ͬ�ĺ��塣��������������һ��ַ������ڶ����ɣ���һ��ֵı�̬������ͬ�� ���塣������߲�����ͬһ���
21. ����˵��� (3-17)ʽ��ÿһ�������������
��(3-17)ʽ����A(l��T��p)����*A(l��T��p)��RTlnxA�֦���A(l��T)��RTlnxA
��A(l��T��p) ��Һ̬��Һ����� A �� T �¶ȣ�ѹ�� p ʱ�Ļ�ѧ�ƣ���*A(l��T��p)�� Һ̬�� A ���¶�T��ѹ�� p ʱ�Ļ�ѧ�ƣ�����A(l��T)��A���¶�T����ѹ��p�� ʱ�ı�̬��ѧ�ơ�����ѹ����Һ̬��ѧ��Ӱ���С����*(l��T��p)���Ƶ��ڦ���(l��T)�� RTlnXA ����Һ����� A �ϴ� A �Ļ�ѧ�Ƶ��½�ֵ��
22. ����������������������۽ṹ�����������кβ�ͬ��
������������۽ṹ������ͬ�����ӻ���������֮����������ȣ������������� �㲻�������������������ͬ�����ӻ���������֮������������ȡ��������� �ĺ������Ϊ��V(���)��0����U(���)��0����H(���)��0����Cp(���)��0������ �����ﲻ������������������
23. �ԱȽ���� B �Ļ�ѧ�����������������������Ĺ�ʽ���к�ͬ�죿
��������������Ϊ ��B(T)������B(T)��RTlnXB
�ڷ����������� ��B(T)������B(T)��RTlnaB
��ͬ�㣺(1)��ѧ��ʽ��ͬ��(2)��̬��ͬ����ͬ�㣺����������ֱ������Ũ�ȣ�B��
���������������û�ȣ�B����ȣ�B����BXB����BΪ���ϵ����
24. ��������ʽ����һ��ѹ�����Ƿ���ȷ��Ϊʲô��
��B��(s��T)�֦�B*(s��T��p)
��B��(l��T)�֦�B*(l��T��p)
��B��(g��T)�֦�B*(g��T��p)
��ǰ���߱Ƚ���ȷ��������ߵ� ��B �� ����B ֮��Ϊ���̣�VS(p��-p)�� ���̣� VL(p��-p)����ֵ�� ����B��Ƚ���С�ɺ��Բ��ƣ�������ʽ����������ʽ���̣� Vg(p��-p)����Vg��VS��Vl��������̲��ܺ��Բ��ƣ��ʵ���ʽ���ܳ�����
������ �� ѧ ƽ ��
1. ����һ�������ϵ���仯ѧ��Ӧ�����ܦ�Gm��(��G/����)T��p �Ƿ��淴Ӧ�Ľ��ȶ��� ����Ϊʲô��
�𣺶���һ�������ϵ���仯ѧ��Ӧ������ ��Gm ��(��G/����)T��p�淴Ӧ�Ľ��ȶ��仯�� �ڵ��µ�ѹ�����£�����Ӧ�������ܵ��ܺͲ����ڲ����������ܺ�ʱ����Ӧ�����Է��� �������ܼ�С�ķ�����С�Ҳ������ϵ��������G���ŷ�Ӧ���Ȧεı仯�����ͣ���ˣ� ��Gm ��(��G/����)T��p��εı仯���ı䡣��һ���棬��Gm��(��G/����)T��p���Ʀ�B��B�� ���ڦ�B����ɣ�B�йأ������ϵ�У���Ӧ�����������һ���淴Ӧ���Ȧεĸı䣬 ��ϵ����ɷ����仯����B�ı䣬��˦�GmҲ�����仯��
2. ���֪��ijһ��Ӧ��ϵ��һ���¶���ѹ���£��䦤Gm��0������ϵ�еķ�Ӧ���Ƿ���ȫ ����ɲ��
�𣺵��¶ȡ�ѹ��һ��ʱ����Ӧ��ϵ�� ��Gm��0��������Ӧ���Է����С�
(1)��Ϊ�����ϵ����Gm��(��G/����)T��p���Ʀ�B��B����α仯���仯�ģ�Ҳ�������ŷ�
Ӧ�Ľ��У���Ӧ���������٣������������࣬��Ӧ�ﻯѧ���ܺͲ��ϼ�С������Ļ� ѧ���ܺͲ����������մﵽ��ȡ���ʱ��Gm��0����Ӧ�ﵽƽ�⡣��Ӧ�������� ��������ʱ����ı䣬��ʱ��Ӧ������ﹲ�棬�ʷ�Ӧ���ܽ��е��ס�
(2)��Ϊ������ϵ����Щ��Ӧ��Gm���Ʀ�B��B����α仯���仯����Ӧ�ﻯѧ��֮���� �Ǵ��ڲ��ﻯѧ��֮�ͣ���˷�Ӧ�ܽ��е��ף����磺��ѹ���£�900��ʱ���ڿ� ��������ʯ��ʯ��Ӧ��CaCO3(s)����CaO(s)�� CO2�������ܽ��е��ס�
3. ���ڷ����ϵ�е����෴Ӧ cC����ġ��� ��G����H
(a)��� ���G�� ���H��c��C�����D
(b)��� ���G�� ���H��c��C�����D
(c)��� �����G�������H��c����C�������D
(d)��� ���G�����H��c��C�����D
�������ֿ�����������Ӧ��ϵ����ʲô�����
��(a) ����Ӧ���Է����� (b) ����Ӧ���Է�����
(c) �μӷ�Ӧ�����ʶ����ڱ�̬ʱ�ﵽ��ѧƽ�⣬���Ǵ��ڱ�̬ʱ����һ�� ��ƽ��̬��
(d) ��Ӧ����ƽ��״̬��
4. ����һ�����µ�ѹ�µķ�շ�Ӧ��ϵ�����䦤G����-RTLnK�����ʷ�Ӧ��ϵ�ı�̬ ����ƽ��̬����������Ƿ���ȷ�������ϵ�����Ϊ��G���� 0������ϵ�Ƿ���ƽ��̬�� ����������ȷ����G����K�����Զ�Ӧ�ڷ�Ӧ��ϵ���ֲ�ͬ��״̬����G����ָ��Ӧ��
����ﶼ���ڱ�̬��������֮�K���Ƿ�Ӧ��ϵ����ƽ��̬ʱ��Ӧ��ֵĻ�Ȼ��� ��G������RTLnK������ӳ�˦�G�� ��K��֮����ֵ�ϵĹ�ϵ����û��������������ϵ�� ��û �б�����ͬһ״̬������ϵ�Ц�G����0����ϵ��һ������ƽ��̬����Ϊ��G����0�� ��ϵ�� ��Gm ����G����RT��nQ��RTLnQa��ֻҪ�����Qa��1����Gm��0����ϵ�Ͳ��� ��ѧƽ�� ̬��ֻ�е���ϵ��Qa��1����Ӧ��ϵ�и���Ӧ���ʶ����ڱ�̬����Gm��RTLnQa��0�� ��Gm����G����0����ϵ�Ŵ���ƽ��̬�������һ������������
5. Ϊʲô��ѧ��Ӧһ��������һ��ȷ����ƽ��̬����Ӧ��ϵ��ƽ�������ǡ�i��i��i��0 ���� Ka��
�𣺶��ڷ����ϵ��˵����ѧ��Ӧһ������һ��ȷ����ƽ��̬����Ϊ�����ϵ��һ������ �£���Gm��(��G/����)T��p���Ʀ�B��B ���ŷ�Ӧ���Ȧεı仯���ı䣬���������ʱ�� ��Ӧ��Ũ�ȼ�С����Ӧ�ﻯѧ��֮�ͽ��ͣ�����Ũ�������ﻯѧ��֮�����ߣ��� ���Ȼ���·�Ӧ�ﻯѧ��֮������ﻯѧ��֮����ȣ����ڷ�Ӧ������ﹲ��һ�� ��Ũ�Ȳ���ʱ����ı��ƽ��̬�������ԭ���Dz����ڷ�Ӧ������һ�����ڻ�ϣ�
�����������ܽ��ͣ�ʹ��ϵ�������淴Ӧ���ȵı仯��������һ����Сֵ����ƽ��ʱ�� ��������͡���ˣ������ϵ��Ӧһ������һ��ȷ����ƽ��̬�� �����ڳ�����ϵ���� �ܲ�����ƽ��̬����Ӧ��ϵƽ��������ǡ�i��B��B��0��������Ka��
6. ��ijһ��Ӧ��ƽ�ⳣ����һ��ȷ������ij���������仰�Ƿ�ǡ����
�𣺲�ǡ����ƽ�ⳣ������ijЩ��Ӧ������(���¶�)�仯���仯��
7. ����������Ӧ��ƽ�ⳣ���к�����(������λ)����G�� ���������ģ�
A���� B �� B���� A nA����(A)n �� ��nA������(A)n
��A����B��B����A��ƽ�ⳣ���������ٶ���Ϊ���������ߦ�G������ֵ��ȣ����� �� ���� nA����(A)n ����nA������(A)n ǰ�ߵ�ƽ�ⳣ���������Ǻ��ߵ�ƽ���� ǰ�ߵĦ�G�� �Ǻ��ߵĶ�����
8. ��Ϊ�����������ܣ�Ϊʲô���ڷǻӷ������ʦ�G����f�֦�Gm��f����������̬�� ������ȴ��ܴ�
�𣺹涨��̬���ȶ���������������Ϊ�㣬�ɱ�̬�µ��ȶ��������ɱ�̬��1mol �Ļ����������������ܵĸı�ֵ��Ϊ�û������Ħ�������������� ��G����f���� ����f G�� ��p��
��(����G/��p)T����Vm�� �� d��G������Vdp�� ��fG������fGm����V(p����p)�� ? ����fGm ��p
��fG������fG*m����Vm(p����p)�����ڷǻӷ������ʦ�Vm ��С����ˣ�
��Vm(p����p)���� ��f Gm ��ȣ��ɺ��Բ��ƣ���fG���֦�fG*m��������̬���ʦ�Vm ��ֵ�ϴ�Vm(p����p)��ܺ��Բ��ƣ���� ��fG���� ��fG*m ���ϴ�
9. Ϊʲô���ڼ���Һ�෴Ӧ�Ħ�Gm����ѹ�������ߵ������£�Ҳ�����æ�*B���������B��
�𣺦�B(L)������B(T)��RTLn��B
��p ��p
�ߦ�*B(l,T,p)������B(l,T,p��)���� VBdp����Һ����ϵ������ ��VBdp��VB(p��p��) ? ��p�� ��p��
��ѹ��p�����������£�����ֵ��С��������������������B�Ǽ�ʮ��ǧ��������ȣ� ��˿ɺ��Բ��ƣ���*B�֦���B�����æ�*B�������B��д�ɦ�B(��)����*B(T)��RTLnaB��
10. ���෴Ӧ�� K����Kp��KC��Kx ��Kf ֮��ʲô��������ϵ���������������٣�
�Ʀ͡Ʀ͡Ʀ͡Ʀʹ�(1)�����������壬K����Kp(p��)-��Kp��K��(p��)��Kx(p)��KC(RT)��
K����Kp��KC �����¶ȵĺ�����Kx ��T��p�ĺ�����K����Kx �����٣�Kp
�Ʀ͡Ʀ� ����Ϊ(pa)��KC ����Ϊ (mol/dm-3)���� �Ʀͣ�0���������١�
�Ʀ� (2)����ʵ�����壬Kf��KpKr��Kf��K��(p��)��K����Kf �����¶ȵĺ�����Kp
�Ʀ� ��T��p�ĺ�����K����Kx �����٣�Kf��Kp ����Ϊ(pa)���Ʀͣ�0���������
�١�
11.�����Ƿ�Ӧ��ϵ��һ���ܽ�����ѧƽ�⡱����������Ƿ�һ����ȷ���Ծ���˵��֮�� �𣺲�һ����ȷ������ڳ�����ϵ�����෴Ӧ�ܽ��е��ף����ܽ�����ѧƽ�⡣���磬 1000�� ʱ�ڿ���������ʯ�ң�CaCO3(s)����CaO(s)��CO2(g)�������Ӧ���ܽ��е� �ף����ܽ�����ѧƽ�⡣
12. �����ӵ��·���ʽ�ó���ѹ����ʽ�����������̸��м�����ʽ����˵��ʲô���⣿ ��(1)���·��̦�Gm����RTLnK����RTLnQa�������ϵ��ÿһ����ֶ����ڱ�̬ʱ�� ����ʽ���Ϊ��G������RTLnK���������������ƽ�ⳣ��������ʽ���κ��¶� ���̿ɵ�����ѹ���̣� ��G��/T����RLnK��
[��(��G��/T)/��T]p��[��R��(LnK��)/��T]p����R[��(LnK��)/��T]p
���������չ�ʽ��[��(��G��/T)/��T]p������H��/T2
�� [��(LnK��)/��T]p����H��/RT2 ��Ϊ��ѹ����ʽ
(2)���·�����ʽ�У���Gm����G����RTLnQa
��G������RTLnK����RTLnQa��RTLn(Qa/K��) �ȡ�
��ѹ���̳��������ʽ (��Lnk��/��T)p����H��/RT2 �⣬���У�
LnK��������H��/RT��C
Ln[K��(T2)/K��(T1)����H��(T2��T1)/RT1T2 ����ʽ��
(3)���·���˵����Ӧ���еķ������(ƽ��)����ѹ����ʽ˵���¶ȶ�K���� Ӱ�죬���¶ȶԻ�ѧƽ���Ӱ�죬�Լ���ͬ�¶�ʱ K��(T) �ļ��㡣
13. Ϊʲô��Ӧƽ����ϵ�г�����������������ϵ��ѹ����Ч��
����Ϊ�ڵ�ѹ�£���ϵ����ѹ��������£���ƽ����ϵ��һ������������壬��ϵ ��������Ħ�������ӣ���Ӧ��ֵ�Ħ���������٣����Ȼ���ͷ�Ӧ��ϵ����Ӧ��ֵ� ��ѹ��������Ҳ�����˷�Ӧ��ֵ���ѹ���������ѹ���������£�������������뽵 ����ϵ��ѹ����ƽ���Ӱ���Ч�����������ѹ���������£����ǵ��������£����� ���������뽵ѹ����Ч�������� �� ƽ ��
1. ͼ 5��2�У��� ������ �� �����ཻ�ģ�������һ���Ȼ�ѧ�Ƶ����ߡ���仰�ĺ��� ��ʲô?
�𣺣���������Tpƽ���ϵ�ͶӰΪ a'd'b'�������ϵ�����һ�㣬����ָ�����¶Ⱥ�ѹ ���£����ѧ����ȣ����� �� �� ���ദ��ƽ��״̬��
2. ���ظ�ԭ����ѹΪ 65.8Kpa��Ϊʲô�����ظ�ԭ��һ����Ӳ��ܽ������ճ��췹�� �𣺲鲻ͬ�¶���ˮ�ı�������ѹ������֪ 89�� ʱˮ�ı�������ѹΪ65.8Kpa�������� ��ԭ�����ȵ� 89�� ˮ�ͷ��ڣ�һ�������ˮ�²��ܳ��� 89�棬�¶Ȳ����ߣ����� ���ײ��ܳ��췹��
3. Ϊ�˷�ֹ����ϩ�ĸ��¾ۺϣ����Բ��ü�ѹ������ʹ����ϩ�� 318.2K ��ѹ���� ��������豸����ն�Ϊ���٣�����㷨��������Щ���ݣ�
�𣺸��ݿˣ����˷��� ln(p2/p1)��(��vapHm/R)(1/T1��1/T2)������֪һ��ѹ��(p1)�� �ķе�(T1)��������(��vapHm)���ݣ��Ϳ����� 318.2K(T2) ʱ����ϩ�ı�������ѹ (p2)����Ϳ�����豸����նȡ�Ϊ�ˣ�����Ҫ֪������ϩ�������е�(��һ���¶��� �ı�������ѹ)��������(��vapHm)��ֵ��
4. ����һ������N�ֻ�ѧ��������ɵ���ϵ������ϵ�в����ڻ�ѧ��Ӧ����N��C����ʲ
ô��ϵ? ��������ijһ�ֻ�ѧ������ijһ����ʵ�ʲ����ڣ����Ƶ�������ϵ��������ʽ ��Ȼ��(5��14)ʽ������ʾ���뿼��������ϵ��ɵı���������������ڸ���仯ѧ�� ��ȵĵ�ʽ��Ӧ������������������
�𣺻�ѧ������N��һָ��ƽ����ϵ���ܹ����ֵIJ�ͬ��ѧ���ʵ��������������C��ָ ��ƽ����ϵ�������������������������ƽ����ϵ����������ȥ��ѧƽ�������ټ�ȥŨ ��������������C��N��R��R���������ϵ�в����ڻ�ѧ��Ӧ����C��N��R���� �����ϵ�в����ڻ�ѧ��Ӧ��Ҳ������Ũ��������������C��N��
C�������ڦ� �����ж����ڣ���������ϵ��ɵı�����Ϊ ��(C-1)����ѧ����ȵĹ� ϵʽ��Ϊ C(��-1)��������ϵ���ٱ������������ɶ� �棽��(C-1)��C(��-1)+2 ��C-�գ�2��
��ij������������ijһ���в����ڣ���������ϵ��ɵı�����Ϊ (��-1)��(C-2) ����(C-1)��1����ѧ����ȵĹ�ϵʽ��Ϊ�� C(��-2)��(C-1)��C(��-1)��1�����ɶ� �棽[��(C-1)-1]��[C(��-1)��1]��2���գ�C��2�������ϵ��������ʽ��ͬһ��ʽ��
5. ���ڴ�ˮ����ˮ����ˮ��������ʱ�������ɶ�Ϊ���٣�������������ģ� �𣺸������ɣ�ˮ�����ʣ�c��1��T��273.16K��p��611pa���¶�ѹ��ȷ���������ɶ� �棽1-3+2��0��˵������ϵ����ɡ��¶���ѹ������������ı䡣
6. ��ͼ 5��6 �У�����ϵ�� a ���ƶ��� c �㣬��ϵ��״̬�����������ı仯��
����� d ���ƶ��� a �㣬���ֽ����������ı仯��
����ϵ��״̬ a �㿪ʼ��������ѹ������CTʱ����ʼҺ������ʱT��p
�����ֲ� �䣬ֱ������ȫ�����Һ���ѹ���ż������ӵ��� c��״
̬����ϵ�� d ��״̬������ѹ���£�����CT��ʱ����ʼ��������ʱ
T��R�����䣬ֱ ��Һ̬ˮȫ�����ˮ�������µ� a ��״̬��
7. ��ͼ 5��8(b) �У�����ϵ�� a ���ƶ��� d �㣺 (a) �����ı䣻
(b) ���ٸı䣬
��ϵ��״̬�����������ĸı䣿
�𣺴� a��d �Ļ����仯���̣�Һ̬���ں�ѹ�½����¶ȣ������C���ϣ�'��ʱ������ �ɵ�б��b'��c' Ϊ��б���ѹ���¹��̣������B����c��ʱ����б��ת��Ϊб�� �˺� c��d Ϊб����ĺ�ѹ����ѹ���̡�
�� a��d �Ŀ��ٸı���̣�Һ̬���ѹ���µ� b'�㣬������ת��ɵ�б���¶Ⱥ� �콵����G���� b ��ʱ����ʼ�����б����b���� Ϊб�����ѹ���¹��̡�
8. ��ˮ�����Һ̬ˮ�Ƿ�һ��Ҫ��������ƽ��̬���Ƿ�������;����
�𣺲�һ��Ҫ��������ƽ�⡣��ˮ����ͼ�Ͽ�֪����ˮ�������£�ʹ�¶ȳ��� 647.2K�� ���ټ�ѹ��ʹѹ������ 2.2?107pa��Ȼ�����Ƚ��º�ѹ����ɲ�������Һ����ƽ�� ̬��ʹˮ��ת���Һ̬ˮ��
9. ͼ 5��9 �� CO2 ����ͼ������ˮ����ͼ�Ƚϣ������к���ͬ��ͼ�����Ĵ� �棽0�� ������ͼ����������ڸ�ѹ��ƿ�ڿ��Դ���Һ̬ CO2������Һ̬ CO2 �Ӹ�ƿ�ڿ����� �������У��������װһ�ش����ڴ��ڵõ����ǹ�̬ CO2(�ɱ�)�������ܵõ�Һ̬ CO2��
����ˮ����ͼ�Ƚϣ������IJ���ǹ�Һ����ƽ���ߵ���б����ͬ��ˮ��Һ�̶���ƽ�� ��б�� dp/dT��0���� CO2 �� dp/dT��0��
�����º���������ѹ��p/p����1ʱ��Һ̬ˮ���ȶ��ģ������� CO2 ��˵����̬���� ̬���ȶ��ģ���Ϊˮ�������ʱѹ�� p/p����1���� CO2 �����ʱѹ�� p/p����1��Һ ̬�Dz����ȶ����ڵģ������̶����ƽ����ڣ���˴Ӹ�ƿ������� CO2 ��������Һ ̬����ѹ��Һ̬ CO2 �����ƿ��ѹ��Ѹ�ٽ��ͣ�Ѹ���������ͣ���ϵ�Ի����������� ���½����¶Ƚ��ͣ�ʹ��һ���� CO2 ���¶��½�������ɹ�̬ CO2(���ɱ�)����� �ش��ڵõ��ɱ��������ܵõ�Һ̬ CO2 ��

10. ���������ͼ���뿼�����������⣺
(a) Ϊʲô������ĸ������?
(b) �������µ�б����Ͷ�� 373.2K �ķ�ˮ�У���Ѹ��ȡ�������ڷ�ˮ�з���һ�νϳ� ��ʱ����ȡ���������ߵ�״̬�����кβ��
(c) �� 388K ���������� 373K �ķ�ˮ�з���һ��ʱ�䣬��ʲô״̬��
(d) �������������ֱ�ͨ�� 373K �ķ�ˮ���� 298K ��ˮ�У����γɵ������ʲô״ ̬?
(e) ����б��Ѹ����ȴ�����£���״̬���ȶ�̬���ǽ���״̬�����ú��ֽ������� ��(a) ��Ϊ����е�б��б�����־��࣬һ��������һ��Һ����࣬���Ծ����� ������㡣
(b) ���� 373.2K ��ˮ��Ѹ��ȡ�������������о���ת�䣬��Ϊб�����ڷ�ˮ�о� �ź��ת��Ϊ��б��
(c) ���Ϊ��б��
(d) ͨ���ˮ��Ϊ��б��ͨ�� 298K ˮ��Ϊб����
(e) Ѹ����ȴ��Ϊ��б�����ǽ���״̬�����ú�ת��Ϊб����
11. Ϊʲô���� 40�� Cd �� Bi��Cd ��ϵ���䲽�����ߵ���״�봿 Bi ���� Cd ����ͬ? ������ϵ��ȴ�� 413K ʱ�����й�����֣��������Ĺ���ɷ���Һ��ɷ���ͬ�� Һ����ɲ��䣬�棽0���¶Ȳ��䣬�������߳���ƽ̨��ֱ��ȫ�����̺��¶Ȳ��� �������Բ���������״�봿 Bi �� Cd ��ͬ��
12. �����ɴӺ� Cd 80�� �� Bi��Cd ������з���� Cd �����ܷ�ȫ����������� ���Ƚ���ϵ�����ۻ�(�¶���� 563K )����ʹ�仺����ȴ��(����ʹҺ����ɾ���)���� ��ϵ��ȴ��BC�ߴ������д� Cd ������������ȴ��Cd ����������ʹ�¶Ȳ����� 413 Kʱ����ɷ��������̬ Cd�������ܰ���ϵ�� Cd ȫ�������������Ϊ�¶Ƚ��� 413K ʱ������ Cd �����⣬Bi Ҳ������Ҳ����Ϊ Cd �� Bi �γɵ��ۻ����(�� Cd40��) �������øܸ˹�������֪������� 66.7�� �� Cd ���Է��������
13. ��ͼ 5��13 �� 5��14 �и��м����棽0�ĵ㣿
����ͼ 5��13 ������� �棽0 �ĵ㣬�ֱ��Ǽ��ᡢ��ȩ���۵�(M����R��)������ ���۵�(p��)�Ͷ�������۵�(N����Q��)��
��ͼ 5��14 �����ĸ��棽0 �ĵ㣬CaF2��CaCl2 ���۵�(A����ĵ�)�����۵� (C��)�Ͳ��ȶ�������IJ�����۵�(B��)��
14. ͼ 5��18 �� 5��19 ��ʾ�����ֲ��ֻ�����ϵ�кλ�����֮ͬ�������кβ�֮ͬ���� �𣺻�����ͬ�������߶����γɶ��ֹ������ �� �£����߾����ж���Һ��ƽ������һ�� ����ƽ���ߡ���֮ͬ���ǣ�ǰ�����ִ�������ʵ��۵�ȽϽӽ�����ϵ���γ�һ��� ���۵㣬�����ߵ����ִ�������ʵ��۵����ܴ���ϵ�����γ�����۵㣬���� ��ת�۵㡣����(��)��Һ����ƽ��ʱ��ǰ�߹�������Һ���ߵ����( Cd ������Һ���� �����ڹ���� ��)�����߹�������Һ���ߵ��ұ�( Cd������Һ����С�ڹ���� ��)��
15. λ��ͼ 5��22 �� A'B' ֱ���Ϸ���ijһ�����DZ�ʾ��ϵ�ĵ㣬������ϵijһ��ĵ�? λ�������ڵĵ�����ϵ�ĵ㣬������ϵ��ijһ��ĵ�? �������Ƹõ������Һ����� ���������
���� A'B' ֱ���Ϸ���ֻ����һ���࣭��Һ�࣬��������һ���DZ�ʾ��ϵ�ĵ㣬Ҳ�DZ� ʾҺ��ĵ㣬������һ�µġ� λ�������ڵ�һ���㣨��ϵ㣩���DZ�ʾ��ϵ�ĵ㣬 ������㣬�����õ����������ƽ���ߣ���Һ���ߺ������߽��㣨M��N�����ֱ��ʾ ����ϵ�У�ƽ�������Һ�������㡣 ���ݸܸ˹�������Һƽ�������������� W(g)��W(l)��OM��ON��
16. ���������ɣ���ͼ 5��22 ���м�飬�� A������ɶ�Ϊ����*��2-2+1��1
��˵����T�Ѻ㶨�⣬ѹ������ɻ��������ɸı䡣��������Ƿ����ʵ������� Ӧ��ξ�����
��A��Ϊ��˫�����ϵ������㣬ʵ������� C��1���ѳ�Ϊ�������ϵ���������� �ɶ� ��*��2-2+1��1������ʵ�������Ӧ�� ��*��1-2+1��0������T�㶨�⣬ѹ�� ����ɾ����ܸı䡣
17. ͼ 5��25 �� 5��26 �е�����ֵ�������ɶ�Ϊ���٣���ε�����˵��ʲô���⣿ ʲô����˫Һϵ����־�ֵ�����ߣ�
��ͼ 5��25 �� 5��26 �е�����ֵ�������ɶ�Ϊ�㣬��*��0�������ķ��������֣� һ���ǣ��ɼ���Ϊ��ֵ����Һ�������ͬ������Ϊ�������ϵ����*��1-2+1��0�� ��һ���ǣ����������Ƶ����ڼ�ֵ��ʱ��Һ���������ͬ��������ϵ��ɱ��������� �ɦ�(C-1) �ı�� (��-1)(C-1)����ѧ����ȵĵ�ʽ�����䣬���ԣ�f��(��-1)(C-1) ��C(��-1)��2��3���� ����ѹʱ f*��2���ա���˲��۵�˫��ֻ����������ϵ�� ֻҪ����ƽ��ʱ�����ͬ�� �����ɶȣ�*��2-2��0��
˵����ѹ�£�����ϵ�¶Ⱥ���ɲ��䣬�Ǻ�л��� ��������ֵ����ַ��Ӽ� ������������ֵ�ͬ�ַ��Ӽ��������ϴ���Һ����ֵ�����ѹ�����ڶ����ɼ��� ֵ�нϴ��ƫ��ʱ���γɵ�˫Һ��ϵ����־��м�ֵ�����ߡ�
18. ����HCl 24�� ����������������������? �������HCl 18�� �������� �������(��� 5��3)��
��HCl��H2O ��ϵ�ĺ�л��������Ǻ� HCl 22.25����������ߺ�е㡣�� HCl 24�� ����������������� HCl����24����ʣ�����е� HCl��С��24����������22.25���� ��� �� HCl 18�� ����������������� HCl����18����ʣ������ HCl�� ����18���� ��С�� 22.25����
19. ����˵�������ϵ��ʵ����ͼ�У���״̬������ɶȾ����������ɶȣ�����Ϊ�Է� Ϊʲô��
�𣺶Եġ���Ϊ�������ϵ�У�C�������棽�����գ�����min��0ʱ���գ����������� �Դ�������ƽ�⡣���� ��min��1ʱ���棽��������˵�������ϵ���������У����� ��������Ҫ����ά������������ر�ʾ����ͼ������ά��ͼ��������ʵ����ͼ����� ��ά����������ͼ�����߶�ά����ƽ����ͼ�����ͨ����ѹ���㶨�����¶ȡ�ѹ������ ������ʱ�����ɶȾ����������ɶȡ�
20. ��ͼ 5��35 ��ʾ����ϵ�����¶��½�ʱ���Դ������ϵ�Ķ�����β��ֻ��������� ������ֱ����������ɴ�״���������ʱ��ͼ�и��������״̬��
��A��B��A��C�Dz��ֻ��ܣ�B��C����ȫ���ܣ����¶��½����γɣ��c���״ (����ͼ)����ʱ��ͼ�ϣ����A����Ǧ� ��(������)�����B��CcΪ �� ��(���� ��)��������c��Ϊ�� �� �� ����ƽ�����������ϵ������ԣϵ��ʾ��M���ʾ
�� ����ɣ�N���ʾ �� ����ɡ�
ͼ 5-35 ͼ 5-36
21. ��ͼ 5��36 �����鲿�ֻ���������ϵ���¶��½�ʱ�������������������� ֱ������ǣ���ͼ��������һ�������Σ��Է�����ϵ�����������е�״̬�� ��ͼ������������� A'B'C'���������� A'B'C' ������һ�㣬
����������ƽ�⣬�������ɣ��� ������� A' ���ʾ��
�� �������� B' ���ʾ���� �������� C' ���ʾ��
22. ������������ͼ�ϣ���һ�ɶ������Ա���һ������ߣ���ͼ 5��37 �е� Cf �ߣ��� �ü��η�֤���ڸ�������һ�㣬A �� B ����ɱ�Ϊһ��ֵ��
��Cf ��������һ��p�������������ϵ�����������ʾ���ص㣬p������ A/B�� Cb/Ac��ap/bp����AB�������ϵ�У������ϵ�� A/B��Bf/Af��Ҫ֤����������� һ����ϵ�����A��B�ĺ���֮��Ϊһ��ֵ����֤�� ap/bp��Bf/Af ���ɡ�
���ݣ��� ��CfB �У�ap��Bf��ap/Bf��Cp/Cf��
��CAf �� b'p��Af��b'p/Af��Cp/Cf�� �� ap/Bf��b'p/Af
�� �Npbb'���Npb'b'��60�㣬bp��b'p
�� ap/Bf ��bp/Af �� ap/bp��Bf/Af

�������ϵ����һ����ϵֹ�����A��B�ĺ���֮��Ϊ��ֵ��(֤��)
������ ͳ������ѧ����
1. ���״̬ȷ����������ϵ���±�����˵������ȷ�ģ�
a.��״̬���� �� ��ȷ��ֵ��
b.ֻ��һ��ȷ������״̬��
c.ֻ��һ��ȷ���ķֲ���
��(a)��ȷ���� S��ln��������ϵ�ĺ��״̬һ��ȷ�����;���һ������ֵ���Ӷ��� ��һ���� �� ֵ��
2. �±߹��ڷֲ���˵������һ������ȷ�ģ�
a. һ�ֲַ�����һ����״̬������ֻ��һ����״̬��
b. һ�ֲַ��������о�������Ϊ ��1 ����һ������n1 ��������Ϊ ��2 ����һ���� ��n2?����������Ϊ ��i ����һ������ni ��
c. ���и��������ĸ�����ӣ�����һ���ʾһ�ֲַ� ��
d. ���ֲַ�������ͬ�ij��ּ��ʡ�
��(b)��ȷ����Ϊ���Ϸֲ��Ķ��塣
3. ���˹ά������������ͳ��ֻ��Ӧ���ڶ���������ϵ���������������һ������ ��һͳ�Ƶ��ص�?
a. ���״̬���� N��U��V Ϊ��ֵ�ķ����ϵ��
b. ��ϵ�ɶ����ɱ�������� U����ini��i��
c. ���ܼ��ĸ�����״̬�з�������������ܰ��ﲻ����ԭ�������ƣ�
d. һ��ʵ�ֵ���״̬������ͬ�ļ��ʳ��֡�
��(c)����������ͳ�ƣ��ܱ��ﲻ����ԭ�����ƣ�����ϵ��Ӧ������ͳ�ơ�
4. ʹ�����˹ά-���������ֲ����ɣ�Ҫ������ N �ܴ�������Ϊ���Ƴ��ö���ʱ�� a. Ӧ������δ�������ӷ���
b. Ӧ����˹������ƹ�ʽ��
c. ����������֮�������ã�
d. �ٶ��������ǿɱ�ġ�
��(b)��ȷ������Ӧ����˹���ʽ������������ N ����ܴ�
5. ����һ������������ϵ�����ܼ��Ϸ����������Ŀ����С�ڸ��ܼ��ϵ��������� �𣺿��ԡ��� M����B �ֲ����� L��K �����ܼ���������֮��Ϊ��
nL/n����(gL/g��)EXp[��(��L������)/KT]
��ʽ�� L Ϊ���ܼ������ڦ�L����������ʽ���ұ�ָ��ʽС�� 1���� ����T����ʱ�� �ӽ���1���� gL/g�� ��1 ʱ������� ���� nL��n����
6. д�����ʵ���Ϊ1Ħ��ʱ��������ϵ���ء����ܡ��ʡ���ķ���������ܼ����� ˹�����ܵ�����ѧ������ͳ������ѧ����ʽ��
��Sm(�ɱ�)��NKlnq��U/T��Rlnq��RT(��lnq/��T)V
Sm(���ɱ�)��NKln(qe/N)��U/T��Rln(qe/NA)��RT(��lnq/��T)V
Um(�ɱ�)��Um(���ɱ�)��RT2(��lnq/��T)V
Hm(�ɱ�)��Hm(���ɱ�)��RT[T(��lnq/��T)V��V(��lnq/��V)T]
Am(�ɱ�)����RTlnq
Am(���ɱ�)����RTln(qe/N)
Gm(�ɱ�)����RT[lnq��V(��lnq/��V)T
]
Gm(���ɱ�)����RT[ln(qe/N)��V(��lnq/��V)T]
7. ���涨����ܼ�����Ϊ ��0������ϵ 0K ʱ������Ϊ U0��N��0�����涨 ��0��0���� U0��N��0��0�����������ϵ���ܵ�����?
���� U��NKT2(��ln��/��T)V �� U��0��NKT2(��ln����0/��T)V
U0��NKT2(��ln��0/��T)V
�� ����0����0EXp(����0/KT) �� U��0��U0��NE0
����ѡȡ ��0 Ϊ����ܼ�������ֵ����ϵ�����ܱ�ѡȡ��������ܼ�������ֵ�� N��0������ϵ�� 0K ʱ������Ϊһ���ģ���ֵ��ѡȡ U��0��Ҳ��ѡȡ U0������ ��� N��0 ��
8. ����ֺ��������壬˼��ƽ����ת��������ֺ����ֱ����¶ȵĹ�ϵ��
���� ��(ƽ��)��(2��mKT)3/2V/h3
��Ϊ�����Һ���� ��(ƽ��)�� T3/2����Ϊ���壬���� V��NKT/p��
�� ��(ƽ��)�� T5/2��
�� ��(ת��)������2IKT/��h ���(ת��)�� T
�� ��(��)����iN[1-exp(-h��/kT)] ���У����ͷ��� N��3n-5 ��
�����ͷ��� N��3n��6���ɼ� ��(��)�� T ��ϵ��
9. Ϊʲô�����Ͷ�ԭ�ӷ��ӣ���ģʽΪ 3n-6�������ͷ�����Ϊ 3n-5��n �Ƿ����� ��ԭ������?
�𣺶���һ�������ԭ����ɵķ��ӣ���Ҫȷ��ȫ�����ӵ�˲ʱλ�ã���Ҫ��n����
�꣬������������Ϊ�������꣬���������ӵ�ƽ�����ɶȣ��������ͷ���Ҫ������ ����Ϊ�涨�˷��������ijһ�̶������꣬�������ͷ��ӵ�ת�����ɶȣ�������� 3n��5 ������ȷ��ԭ�Ӽ�����λ�ã����� 3n��5 ��ת�����ɶȡ����ڷ����ͷ� �ӣ���Ҫ��������ȷ����ռ�ȡ������ת�����ɶȣ��������ɶ�Ϊ 3n��3 ��3��3n��6��
10. ���͵�ԭ�ӷ����������� CV��m����R��˫ԭ�ӷ�������������ͨ���¶��� CV��m��2.5R���¶ȸ�ʱ���ܵ���3.5R��
�𣺶Ե�ԭ�ӷ��ӣ���0(����)��(ƽ��)����0(����)(2��mKT)3/2V/h3
CV��m��(��Um/��T)V
�� Um��RT2��[��lng0(����)��2��mKT)3/2V/h3]/��T��V��3RT/2
ƽ�����ɶ��У������ɶȣ�ÿ�����ɶȶ� Um ���� RT/2��
�� CV��m��(��Um/��T)V��3R/2��ÿ�����ɶȶ� CV��m ���� R/2��
����˫ԭ�ӷ��ӣ�
��0 (����)?{(2��mKT)3/2V/h3}{8��2IKT/(��h2)}[1-exp(-h��/kT]-1
���룬Um��RT{2.5��(h��/kT)/[exp(h��/kT)-1]}
�����£�exp(h��/kT)-1��exp(h��/kT)��Um��RT[2.5��(h��/kT)/exp(h��/kT)]
Cm��V��(��Um/��T)V��5R/2��R(h��/kT)2exp(h��/kT) ���� h��/k����T��
CV��m����R/2�� ������ƽ������ת�����ɶ��е�ÿһ�˶����ɶȶ� Cm��V Ϊ R/2�� �����£��� h��/kT����T��Um��7RT/2 ���� CV��m��7R/2
���ܹ�������ƽ��������ת����һ�������ɶȣ�ÿ�������ɶȹ���Ϊ CV��m��R��
11. ˼��һ�����Թ�̬��Һ̬����̬����ֵ�仯�������
�𣺿�������������п���
(a) ��ͬһ���� ��(��) �� ��(Һ)�� ��(��)���� S��Kln��
���� S(��) �� S(Һ) �� S(��)
(b) �� S��NKlnq��U/T ��ͬһ������ �� U(��)�� U(Һ)�� U(��)
q(��) �� q(Һ)�� q(��)������ S(��) �� S(Һ) �� S(��)
12. �Ƚ�ͬһ����� Cm��c(ƽ) �� ��c2(ƽ) �Ĵ�С��
��Cm��(2KT/m)1/2 �� c(ƽ)��(8KT/m��)1/2 ��
��c2(ƽ)��(3KT/m)1/2
�� Cm �� c(ƽ)��[c2(ƽ)]1/2
13. һ���������Ϊ V�������� n ������Ϊ m ������������ӡ����ǴӸ��������� ײ���ڶ�����ѹ������ x ����������ײ��������ѹ��ӦΪ���º�ʽ��
a. p��2mnv2x/V�� b. p��2mnc(ƽ)/V��
c. 2mnvx2(ƽ)/V d. 2mnc2(ƽ)/V
��(c)����ȷ�ġ�
14. ���³�ѹ�£�������ӵ� �ơ�10-10 m��n��1024 m-3��c��102 m?s-1�����
�� ZA��ZAA ��ƽ�����ɳ̵���������
�⣺ZA����n��2c��3.14?1024?(10-10)2?102��106��-1
ZAA��(��2)��2n2c/2��(��2)��?(10-10)2?(1024)2?102/2 ��1030 m-3?s-1
L��(��2)��nA��A2/2��c/ZA��10-2/106��10-4 m
15. Ϊʲô�õ���6-136��ʽ�� ZAA �ļ���ʽʱ����2�����õ���6-138��ʽ�� ZAB �ļ� ��ʱ������2��
��(6-136) ʽ��ͬ�ַ��� A ֮�����ײƵ�ʼ��㹫ʽ����ÿ�� A ������ײ�뱻ײ�� ��������ظ���һ�Σ���Ҫ����2���� ZAB Ϊ���ֲ�ͬ���Ӽ����ײ������ A ���� ��ײ B ���ӣ���B������ײA���ӣ�δ�ظ����㣬�ʲ�Ҫ����2��
16. ˼�������ȷ�����һ��ѧ��Ӧƽ�ⳣ���ij���
���� ��G������RTlnK��
�� ��lnK����(1/R)?(��G��/T)��(1/R) ����(G����U����0)/T�ݣ�(��U����0/T) ��ʽ�е� ��U����0 �������ȷ���ã���˼��㻯ѧ��Ӧƽ�ⳣ���ij����裩Ϊ�� (a) ��Ϊ����֤�� ��U����0����H����0
��T
(b) ��H����0(T)����H����0���� �Ʀ�BCp��m(B)dT
��0
(c) ����Cp��m���ᣫ��T��cT2��??
�Ʀ�BCp��m(B)���Ʀ�BaB���Ʀ�BbBT���Ʀ�BcBT2��??
(d) ��H����0����H��(T)���Ʀ�BaBT�����Ʀ�BbBT2�����Ʀ�BcBT3��??
(e) ���㼪��˹�����ܺ����G������ɰ���ʽ�� K����
17. ˼���ù�������һ��ѧ��Ӧƽ�ⳣ���ij���
���� ��G������RTlnK���� ��U����0����H����0 ������������£�
(a) ��lnK����(1/R)?(��G��/T)��(1/R)?����(G����U����0)/T�ݣ���U����0/T
(b) H����T��U����0��(U����T��U����0)��RT��RT2(��lnq��0/��T)V��RT
(c) ���ù�����������ʺ����� (��Ϊ��H����T��U����0 ��)
(d) �Ӷ��ó� ��U����0����H����T����(H����T��U����0)
(e) ���빫ʽ����� ƽ�ⳣ�� K����
���ڡƦ�B�� 0 �ķ�Ӧ�������ù�ʽ��
K�����ۡ�q��B��0(����)/��q��B��0(��Ӧ��)��?EXp(����U����0/RT)
3/2 ��{��MB(����)/��MB(��Ӧ��)}?{��IB(����)/��IB(��Ӧ��)}
?{�Ǧ�(����)/�Ǧ�(��Ӧ��)}?{��q0(������)/��q0(����Ӧ��)
?EXp(����U����0/RT)
��������������������� ��U����0 �⣬��Ҫ�ù���������� IB �����ʵ���
��Ƶ�ʦ�B ��
������ ��ѧ����ѧ
1���Ժ��ݷ�Ӧ aA �� bB���� eE �� fF���䷴Ӧ���ʿ��������κ�һ�����ʵ�Ũ����ʱ��� �仯�ʱ�ʾ������֮��Ĺ�ϵ��( )��
(1) ��a(dcA /dt)����b(dcB /dt)��e(dcE /dt)��f(dcF /dt)
(2) ��dcF /fdt����dcE /edt��dcA /adt��dcB /bdt
(3) ��fdcA /adt�� ��fdcB /bdt�� fdcE /edt ��dcF /dt
(4) dcA /dt��bdcB /adt��edcE /adt��fdcF /adt
��(3)
2��������һ����Ӧ������˵���У�����ȷ����( )��
(1) lnc ��ʱ�� t ��ͼ��һ��ֱ�ߣ�
(2) ��˥���뷴Ӧ����ʼŨ�ȳɷ��ȣ�
(3) ͬһ��Ӧ���ķ�Ӧ��İٷ�����ͬʱ�������ʱ����ȣ�
�� (4) �ٶȳ����ĵ�λ��(ʱ��)1��
��(2)
3��������Ӧ 2A����B �İ�˥����( )��
(1) �� A ����ʼŨ���أ� (2) �� A ����ʼŨ�ȳ����ȣ�
(3) �� A ����ʼŨ�ȳɷ��ȣ� (4) �� A ����ʼŨ��ƽ���ɷ��ȡ�

��(3)
4����֪ij��ӦΪ n ���������ʷ���ʽΪ �� r��kcn
�Ծݴ˵������йض���ѧ����ʽ�������۵� n��0��1��2��3 ʱ�����Ρ�
������dc/dt��kcn ��dc/cn��kdt �����ֺ�ã�[1/(n��1)][1/(cn1)��1/(c0n1)]��kt
�� n��0 ��c��c0��kt c��c0��kt (Ϊ�㼶��Ӧ����ѧ��ʽ)
���������� �� n��1 k1t��(n��1)1[(cn1)1��(c0n1)1] �ұ���0/0 ��
������ ��ֵ����x �� n��1 k1t��Lim x��0 x1[(cx)1��(c0x)1]
�������� ��Lim x��0 (c x��c0 x)/x��Lim x��0 (��c xlnc��c0 xlnc0)/1
���� ��Lim x��0 (c0 xlnc0��c xlnc)��ln c0/c
����k1t��lnc0/c (һ����Ӧ����ѧ����)
�� n��2 (1/c)��(1/c0)��kt (������Ӧ����ѧ��ʽ)
�� n��3 ��[(1/c2)��(1/c02)]��kt (������Ӧ����ѧ��ʽ)
5���ڻ�Ԫ��Ӧ�У���ȷ����( )��
(1) ��Ӧ�����뷴Ӧ����������һ�£� (2) ��Ӧ�������Ǵ��ڷ�Ӧ��������
(3) ��Ӧ��������С�ڷ�Ӧ�������� (4) ��Ӧ������һ���뷴Ӧ����������һ�¡� ��(4)
6����������ʽ A��B ����2p������ȷ��������( )��
(1) �˷�ӦΪ������Ӧ�� (2) �˷�ӦΪ˫���ӷ�Ӧ��
(3) �˷�ӦΪ��Ԫ��Ӧ�� (4) �˷�Ӧ�����ʼ�ļ�����ϵ�Ѷ��� ��(4)
7���������ö��ɶ����ܷ�ӦʽΪʲô��һ����ȷ?
�����������ö���ֻ�����ڻ�Ԫ��Ӧ�����ܷ�Ӧ��һ���ǻ�Ԫ��Ӧ����˲�һ����ȷ��
8������ƽ�з�Ӧ������������ȷ����( )��
(1) k1��k2 �ı�ֵ�����¶ȶ��䣻
(2) ��Ӧ��������ʵ�������ƽ�еķ�Ӧ����֮�ͣ�
(3) ��Ӧ���� B �� D ����֮�ȵ�������ƽ�з�Ӧ������֮�ȣ�
(4) ��Ӧ�����ĵ�������Ҫ�����ڷ�Ӧ���ʽϴ��һ����Ӧ��
��(1)
9���ɴ�A��ʼ�Ķ��ŷ�Ӧ���ڶ����½��У�����˵���в���ȷ����( )
(1) ��ʼʱ��A�������������
(2) ��Ӧ���еľ���������������Ӧ����֮�
(3) ����������ʳ���֮���Ƕ�ֵ��
(4) ��ƽ��ʱ������������ʳ�����ȡ�
��(4)
10����֪���ӷ�Ӧ������Ϊ�����[cA]/dt����( )
(1) k1[cA]��k��1[cB]��k2[cA][cB]
(2) ��k1[cA]��k��1[cB]��k2[cA][cB]
(3) ��k1[cA]2��k��1[cB]��k2[cA][cB]
(4) k1[cA]2��k��1[cB]��k2[cA][cB]
��( �����ԣ�ӦΪ ��k1cA2��k��1cB��k2cAcC )��
11��H2 �� O2 ��Ӧ����ը��ԭ����( )��
(1) ����������������Ӧ�� (2) ֱ�����ݵ��������ӣ�
(3) ���ɻ��������� (4) ����˫���ɻ��γ�֧����
��(4)
12��H2 �� O2 ��Ӧ���ڱ�ը��ѹ�����ޣ���Ϊ( )��
(1) ��������������������٣� (2) �������ʵ�Ӱ�죻
(3) ����������������ײ�����٣� (4) ��Ӧ��̫�ٲ�������Ӧ��
��(3)
13�������ȱ�ը��Ӧ��������ϵ��ȷ����( )��
(1) ��ʼ��Ӧ���� �� �м䷴Ӧ���� �� ���˷�Ӧ���ʣ�
(2) �м䷴Ӧ���� �� ��ʼ��Ӧ���� �� ���˷�Ӧ���ʣ�
(3) ���˷�Ӧ���� �� �м䷴Ӧ���� �� ��ʼ��Ӧ���ʣ�
(4) ���˷�Ӧ���� �� ��ʼ��Ӧ���� �� �м䷴Ӧ���ʡ�
��(3)
14��Arrhenius����ʽ������( )��
(1) ��Ԫ��Ӧ�� (2) ��Ԫ��Ӧ�ʹַǻ�Ԫ��Ӧ��
(3) ���ŷ�Ӧ�� (4) ���л�ѧ��Ӧ��
��(2)
15������ʱ���� van��t Hoff ����Ļ�ѧ��Ӧ�����ܷ�Χ��( )��
������ (1) 40��400 kJ?mol1�� (2) 50��250 kJ?mol1�� (3)100 kJ?mol1��
��(3)
��16��ij��Ӧ�Ħ�H��100 kJ?mol1��������( )��
���� (1) ��С�� 100kJ?mol1�� (2) �ش��� 100kJ?mol1��
���� (3) �ɴ��ڻ�С�� 100kJ?mol1�� (4) ֻ�ܵ��� 100kJ?mol1��
��(2)
17��ָ�����з�Ӧ�ĸ��Ļ����С�����밴���ܴ�С��һ˳��
(1) Cl��Cl��M����Cl2��M�� (2) HI��C2H4����C2H5I��
(3) H��CH4����H2��CH4�� (4) N2��M����N��N��M��
�𣺣�1����Ӧ�����С����С�������Ϊ (1)(3)(2)(4)��
18�����巴Ӧ��ײ���۵�Ҫ����( )ȫ����ӿɿ����Ǹ��� (1) һ����ײ����Ӧ��
(2) ��һ�������Ϸ�������ײ����������Ӧ�� (3)����ӭ����ײ�����ܷ�Ӧ��
(4) һ�Է��Ӿ����㹻��������ײ����������Ӧ��
��(4)
19����ѧ��Ӧ�Ĺ���״̬������Ϊ( ) (1)��Ӧ���ʾ����ڻ�������������ʣ�
(2) ��Ӧ���ʾ����ڻ�����ֽ�Ϊ����ķֽ����ʣ�(3) ������ѧ��������������ʳ�����
(4) ������Ͳ����ɽ���ƽ�⡣
��(2)
20���¶����ߣ���Ӧ����������һ�������ѽ�����( )
(1) ��Ӧ���ӵ���ײƵ�����ӣ� (2) ���ܻ����ݽ��ͣ�
(3) ����������ܵķ��ӵİٷֺ������ӣ� (4) ��Ӧ���̸ı䡣
��(3)
21����Ϊ��������ż��? �ݴ��������Һ��Ӧģ������?
�𣺺���̬��ȣ�Һ̬�����ǽ������еģ���Ӧ�������Һ̬�ܼ����Ȼ������Χ�ܼ�����
�Ľ��ܰ�Χ�С����ǰ�����״̬����ر���Ϊ������������Ϊ��Ӧ��������ܼ��е��˶���
���ڽ��ڷ�������ɵ����е�һ�������ǰ���һ�����еķ�����ײ����Ϊһ��ż������
Һ�з�Ӧ��ģ��Ϊ��A��B����A��B����p��A��B�γ�ż���ԣ��ù��̿��ٴ�ƽ�⣬ż���� �ֽ�ɲ���ù���Ϊ�����̡�
22���ܼ��Է�Ӧ���ʵ�Ӱ�죬��Щ�����������ã������������ʲô?
���ܼ���糣����Ӱ����������ЧӦ���ܼ���Ӱ�����ڻ�ѧЧӦ������ǿ�ȵ�Ӱ����л�ѧЧӦ Ҳ������ЧӦ��
23������Ӧ�ⷴӦ�к�����? ���⻯ѧ��Ӧ�к�����?
������Ӧ�������ǣ���Ӧһ����ʼ�£��粻�ӿ��ƣ��ͻ��Զ��ط�չ��ȥ���䷢չ��ʽ��������һ����
�������������ǹ��գ�Ҳ�����Ǽ��Ȼ�������������⻯��Ӧ���ڿɼ���������¼�����Ӷ����� ��ѧ��Ӧ���������ķ�Ӧ����Щ���ܴ���������Ӧ���� H2��Cl2���������⻯��Ӧ�Ħ���С���Ƿ�
����Ӧ�����⻯ѧ��Ӧ�����ڸ�ǿ�ȵ�ɫ��( ���� )�����䣬�����Ƶ���뻯������ijһ������ Ƶ����ƥ�䣬�Ӷ�ʹ���ض��ļ��������ѣ����������е����������ϣ�ʹ������ͨ�������ܽ��е� ��Ӧ���õ����С�
24�� Franck��Condonԭ������������ʲô? ���о��⻯ѧ��Ӧ�к�����?
����Franck��Condonԭ���������˫ԭ�ӷ��ӵ�������(Լ1013��)���Ե���ԾǨ����ʱ�Ǽ��̵�
��(Լ1015��)�����ڵ���ԾǨ��˲�䣬ԭ�ӵĺ˼�������Ϊû�б仯���ӻ�̬��0�����ܼ�
����0������̬��1�����ܼ�����0 ��ԾǨ���� 0��0 ���ļ������������ǿ��������� ԾǨ����0��1��0��2 ���������������һԭ���ɽ���Ϊʲô���������������Щǿ����Щ���� �Ӷ������Ʋ���ЩԾǨ��ʹ���Ӳ���ǿ����������⡣
25���Ƚ�ӫ��������ͬ��
��ө����ⶼ����⣬�ӵ���̬����̬(S*����S)��ԾǨ�����Ĺ�Ϊө�⣬������Ϊ�����̣�
ǿ��ǿ�����������̣�������̬����̬(T*����S)��ԾǨ�����Ĺ�Ϊ�⣬������Ϊ��������
ǿ������������������
26������������Щ������ ø����һ����к���ͬ��
�𣺴�����������
1. �������뷴Ӧ�����м���
2. ��������ѧ��Ϊ�����Է����еķ�Ӧ������ֻ�����̴ﵽƽ���ʱ�䣬���ܸı�ƽ��λ�á�
3. �����ı䷴Ӧ;�������;������ʲ���Ļ�ܡ�
4. ��������ѡ���ԡ�
ø����һ����Ƚϣ���ͬ��Ϊ���߶�רһ�ԣ��߶ȴ����ԣ�������¶�ЧӦ ��
27������Ϊʲô����ı仯ѧƽ��λ��?
����Ϊ��������ѧԭ������Ӧ���Ƿ�Ӧ��ϵ�ij�̬����������̬��ʼ��̬�Ѿ�ȷ������״̬�����ı�
ֵ ��Gm����G��Ϊ��ֵ���������ܸı�ʼ����̬��Ҳ�Ͳ��ı䦤G�������ı�ƽ��λ�á���֮ ����ı�ƽ��λ�ã�����Ƴ��ڶ�����������Υ������ѧ�ڶ����ɡ�
28�������ʹ���ʲô��ϵ?
�𣺶�����ڱ����Ͻ��У�����������DZ��淴Ӧ�ı�Ҫ������
29�����������ͻ�ѧ������ʲô����? ��ʲô����£����߿����ת��?
�𣺻�ѧ���������������Ƚϣ����ȴ���ѡ���ԣ������Ӳ㣬������ܽϸߣ��ڽϸ��¶��½��С�
���������������ĸı䣬�ر����¶����ߣ���Щ����������ת��Ϊ��ѧ����������ѧ�������ǰ� ������������
30���ɸ��Ѷ�����ʽ���Ƶ���Ӧ����ʲô����?
�𣺱����ǵ������������˵������⣬��ʽ�����ڦȽ�С��������ȣ����������Ӽ�û������� �ĵ����Ӳ�������
31����ѧ����ѧ�ͻ�ѧ����ѧ������������кβ�ͬ? ����˵����
�𣺻�ѧ����ѧ���ijһ��Ӧ��һ�������ܷ���Է����е��оݣ����е�ʲô�̶�Ϊֹ����ѧƽ��̬����
������ѧȴҪ�����Ӧ���еÿ����뷴Ӧ����������ijһ��Ӧ��һ���¶��¾���һ��ʱ��ת����Ϊ ���٣�����ѧ��������ԣ�����ѧ�����ʵ�ԡ����磺�ϳ�NH3��Ӧ��H2���2��ȼ��Ӧ��
32���ϳɰ�����������ΪʲôҪѡ����£���ѹ��ѹ��ѡΪ(1��3)?104 kpa���¶�ѡΪ723��823K�� �𣺺ϳɰ� N2 ��3H2 ����2NH3 �Ƿ��������ٵķ�Ӧ������ѹ���������ת���ʡ��ϳɰ��� Fe ������
Fe ����Ҫ�ڸ��²��л���� 723��823kʱ Fe���Դ��ڽϸ��¶��£���Ӧ�죬��λʱ���� ���ʸߡ�
33�����Ṥҵ�У����������ת����ΪʲôҪ�����Ķ�ת��?
���� SO2����O2 ���� SO3 ��H��0�����ȷ�Ӧ
����ƽ���ƶ�ԭ���������¶�������ƽ�������ƶ������ SO2��ת���ʣ����¶ȵ��ֻ������Ӧ���ʣ� �����ҵ�����в���������һ�δ��¶���ߣ��Ա���ٴﵽƽ��Ȼ������¶Ƚ��μ��͵ĵڶ��Ρ�
�����Ρ����Ķδ��Σ�ʹƽ��λ���������ƶ����Ա���� SO2��ת���ʣ�������װ���ܴﵽ�ȿ죬 ת�����ָߵ�Ŀ�ġ�
�ڰ��� �������Һ
1���¶�����ϣ���鹫ʽ���������ͷ�Χ��ʲô���¶�����ϣ���Ӷ����˶����ɵ���Ҫ�Ժ��ڣ�
�𣺿¶�����ϣ���鹫ʽ��?m????Ac�� ������ǿ�����ˮ��Һ��Ũ�ȵ���0.01mol��dm-3��
ϡ��Һ���������Ӷ����ƶ����ɣ����Դ���ص�ǿ����ʵĦ���������������ʵĦ��ޡ��������� �絼��ֵ���������ʵ�����ϡ��ʱĦ���絼��
2���絼����Ħ���絼�����кβ�ͬ? ���Ǹ�����Щ�����й�?
�𣺵絼�ʦ��ǣ����������Ϊ1m2�������1mʱ�������Һ���ʵĵ絼����Ħ���絼�������
1m�����缫�京��1mol���ʵ���Һ���ʵĵ絼��Ħ���絼�æ�m��ʾ��m����/c���絼�ʦ���� ���ʱ����йأ����¶��йأ�������Ũ���йأ�Ħ���絼�����ʱ����йأ����¶��йأ� ������Ũ���йء�
3��Ϊʲô�ý������Ųⶨ��Һ�ĵ絼? Ϊʲô��1000Hz(��c/s����ÿ��)Ƶ�ʲⶨ��Һ�ĵ絼? Ϊ
ʲô��δ֪�������·�ϲ���һ����? ����Һ�絼�Ĺؼ���ʲô?
���ý�����������Һ�ĵ絼�����Ա��������ö��ı�缫���ԣ����ҿ��������缫�ļ������á�
��1000Hz�Ľ���Ƶ�ʿɷ�ֹ�缫�ϵļ������ã������ö������㡣����������Ϊ�������絼�� �ĵ��ݵ�Ӱ�졣���絼�Ĺؼ����ڸ��Ӵ�����Ӵ��������£�����ƽ�⣬��ȷ���㡣
4����һ��ֱ����ͨ��һ���н������ӵ���Һʱ���������������������������ڣ�
(1) �����ı������ (2) �������Һ��Ũ�ȣ�
(3) ͨ��ĵ����� (4) �������Һ������Ǩ�Ƶ��ٶȡ�
��(3).
5���ڽ����ƶ����ⶨ����Ǩ������ʵ���У������Ƿ���ȷ����ؼ��Ǿ����ڣ�
(1) �����ƶ��������̶ȣ� (2) ��ӵ�ѹ�Ĵ�С��
(3) ����������Ǩ���ٶ��Ƿ���ͬ�� (3) ���������ӵļ����Ƿ���ͬ��
��(1)
��6���������ˮ��Һ��ʱ����Ϊ�ܼ���ˮ����Ϊ H+��OH���ӣ�Ϊʲôһ�㲻�������ǵ�Ǩ����?
Ӱ������Ǩ��������Ҫ������ʲô��
��������Ϊˮ��H+��OH��Ũ�����ͣ�Ksp��1014����Ǩ������С�������Dz���Ӱ����������
����Ǩ�������¶ȼ�Ũ���йأ����Ǹ���Ҫ�����������ܼ����Լ��γ������ӵ�Ӱ�졣
7������ʵ�Ħ���絼�ɿ�������������Ħ���絼֮�ͣ���һ��������������������Һ��
(1) ǿ�������Һ�� (2) ���������Һ��
��(3) ����ϩ��Һ�� (4) Ħ��Ũ��Ϊ1 mol��dm3����Һ��
��(3)
8��(A) ������ʵļ���Ħ���絼Ϊʲô�������Ʒ���ã����ü���ķ�����ã�
ǿ�������Һ�ļ���Ħ���絼Ϊʲô���������Ʒ���ã���ԭ���д����Ըģ�
(B) ���п����ĸ�����ȷ��
(1) ���������Һ�����ֵ��룬�䦫mֵ��Ũ�ȱ仯�Ƚϴ�
(2) ���������Һ�Ħ��������Ʒ���ã�����ȷ��
(3) ǿ�������Һȫ�����룬Ũ�ȶԦ���Ӱ�첻���硣
�⣺(A) ����������ʵ�Ħ���絼��Ũ�ȵĹ�ϵʽ�������ӿ¶�����ϣ���鹫ʽ����ϡ��Һ�в�
�����Թ�ϵ����˲��������Ʒ���ü���Ħ���絼�������Ը������Ӷ����ƶ����ɣ�������á� ��ǿ�����ϡ��Һ���ӿ¶�����ϣ��ʽ�������Թ�ϵ�����������Ʒ��������Ħ���絼��
(B) (3)�Ƚ���ȷ��
9���°ݣ��ȸ������ǿ�������Һ�����ӷ�ģ�ͼ��䶨�������Ļ����������ļ��㣿
�����ӷջ������裺(1) ǿ�������ˮ����ȫ���롣(2) �������Ӽ�����ö��γ����ӷա�
(3) �������ӵ����˶������ӷղ��Ǿ�ֹ�ģ����ӷ���ʱ��ͳ�Ƶ�ƽ�������
���������Ļ������裺(1) ���� Boltzman�ֲ����ɴ������ӷ������ӵķֲ���
(2) ���ò��ɷ����Ƶ���������ѡ����ij�������Ӵ��ĵ��� ?��
(3) ���빫ʽ�еģ����䵹�� 1/���൱�����ӷյ�ƽ�����(���ӷհ뾶)��
10���°ݣ��ȸ����ʽΪʲôֻ��������ϡ��Һ��������÷�Χ��ô��?
����Ũ�Ƚϴ�ʱ������֮�������ǿ��������������Ӽ�Ľ�ϣ�����ʽ���������ϴ���
������÷�Χ�İ취�ڼ���ʽ���һ��CI�������������õ�Ũ�ȷ�Χ��CI��������Ӷ̳�����á�
11���о��������Һ����ƽ�����ϵ�����������Ҫ�Ժ���? �ǵ������Һ�Ļ��ϵ������
�ù�ʽ������
��������Һ�������������������ڣ����ܵ�������������ӵĻ�ѧ�ơ���Ⱥͻ��ϵ����
����ƽ�����ϵ���ĸ����ɼ�������ǿ����ʵĻ�ѧ�����ȡ������á��� = ��+��+ ��-��- ���ڷǵ���ʣ� ��ͣ����ͣ� ��Ϊ�㣬��á�=1���ʷǵ���ʻ��ϵ�������ø�ʽ����������������Է��������
12������ǿ�ȸ�������������ʲô��Ҫ����?
������ǿ�������ӵ�����γɾ��糡ǿ�ȵ����ȣ�������֮�侲�����ô�С�����ȡ�
13�����������ƽ�����ϵ���Ĵ�С��Ҫ�������к������أ�
(1) ����֮��ľ���������(2) ����������ǿ�ȣ�(3) ��������ʵ��������ӵ�ƽ��ֱ���� ����Ϊ��Ҫ������ʲô���ء�
��(1)
14���°ݣ��ȸ�����ۺ����������۸��Խ����ʲô���⣿������������˼����ʲô�����ߵ�
��ϵ��ʲô��
�𣺵°ݡ��ȸ�����۽������μ���ǿ�������Һ�����Ӻ͵���ʵĻ��ϵ���ȣ�������
�����۴������Ͻ����ǿ����ʵ�Ħ���絼�������⣻�������۵�����˼�붼��ǿ������� ��ȫ����ģ����γ����ӷա���ǰ���Ǽٶ����ӷ�ֻ�������˶��У������ζԳƵģ������� ��Ϊ���ӷ��ڵ糡�ж����ƶ�ʱ�����������ζԳƣ��Ӷ�������Ӿ�����ɳ��������������� ���ڵ°ݣ��ȸ�����ۻ����Ϸ�չ�����ģ����߶���������ǿ�����ϡ��Һ��
15���°ݣ��ȸ���������������ڵ�����ϵʽ�п������������أ�������һ������û�п��ǣ�
��Щ���ؿ�����?
(1) �������Һ��������Һ��ƫ����Ҫ�ɾ��������£�
(2) ÿ�����Ӷ�������෴����������Χ��(3) ÿ�����Ӷ����ܼ�����
(4) ����֮������ŵ�����
��û�п��ǵ�������(4)�����ǵ�������(1)��(2)�� (3)
16��������?????A���������������е���һ����
(1) ������ʣ�(2) ǿ����ʵ�ϡ��Һ��(3) ����ϡ��Һ��(4) Ħ��Ũ��Ϊ1����Һ�� ��(2)
17������Щ���ݿ��Ա������ӵ�ˮ�����о�����ˮ����ʲô�������壿
���ɦ�H(��)������H(��)����H(ˮ��)�����ݣ�����H(ˮ��)����H(��)����H(��))��0ʱ������
Щˮ����������ɫ��������ȷ����ˮ��������Щ���Ա������ӵ�ˮ����������ˮ���ĸ���� �Խ���һЩʵ�����⣬�磺�������ʵ��ܽ�����¶ȵĹ�ϵ��ͬһ�ֵ�����ڲ�ͬ���ܼ��� ���ֵ�ǿ����ͬ��ˮ�����ӵķ��֣���������ѧ��ijЩ�������µ���ʶ��
�ھ��� �� �� �� ��
1����Ӧ�õ��Ʋ�Ʋⶨ��ص綯�Ƶ�ʵ���У�����ʹ�����к��ֵ�ػ���Һ:
(1) ����缫��ɵ�أ� (2) �ʹ��缫��ɵ�أ�
(3) ���Ϊ 1 �ĵ������Һ�� (4) ����أ�
��: (4)
2���ڵ��Ʋ����AB�߶�Ϊ���裬Ϊʲô�ܱ�ʾ���Ƶ���ֵ��?
��: ����ŷķ���� E��IRAB��I��AB����ʽ��IΪͨ����·�ĵ�������Ϊ���ߵ�λ���ȵĵ��覸/m��
���ⶨES��Exʱ����ֵ��ȣ�I���ʾ�Լȥ������ ES��AB��Ex��AC������ڵ��Ʋ���п��� �� ES��AB��Ex��AC ����ʾ������������²����õ��߳�������ʾ���ơ�
3���õ��Ʋ�ⶨ��ص綯��ʱ,�����ּ�����ʼ��ƫ��һ��,���ܵ�ԭ��:
(1) �����Ʋ��飻 (2) ���ⶨ�ĵ�ص綯��̫��
(3) ���費���,���Ũ�Ȳ����ȣ�(4) �����ص������缫�ӷ��ˡ�
��: (4)
4��Ϊʲôѡ��Τ˹�ǵ����Ϊ�����?
��: ��ΪΤ˹�ǵ���ܱ�֤�ڲ��������д��ڿ�����̵����������Ҹõ�صĵ綯���ȶ���
�¶�ϵ����С��
5������ں��¡���ѹ�Ϳ��������·ŵ磬�����뻷������Ƚ���Ϊ���к���:
(1) һ��Ϊ�㣻 (2) Ϊ ��H��
(3) Ϊ T��S�� (4)�� ��H �� T��S ���ء�
��: (3)
6�����¶�Ϊ T ʱ������������Ӧ
���� ?Cu��?Cl2��?Cu2��Cl ?1��
���� Cu��Cl2�� Cu2��2Cl ?2��
?1�� ��?2�� �Ĺ�ϵΪ���к���:
(1) ?1����?2���� (2) 2?1����?����
(3) 4?1����?2���� (4) ?1����2?2����
��: (1)
7���ź� ������ʲô�����й�? ������ǿ�����ػ�����������? E��ֵ������һ����ѹ����
�����ʵĻ��Ϊ 1 ʱ�ĵ綯��,��仰����Ϊ����?
��: E �� E����ǿ�����أ��� nEF������G,�� E ���ط�Ӧ�������ڷ�Ӧ�����еĦ�G�йأ�
����G ���¶��йأ��� E ���¶��йء�E�� ������һ����ѹ���£������ʵĻ��Ϊ 1 ʱ �ĵ綯��,��仰ԭ���϶Եģ���������������Ҳ����Ȼ����������ʵĻ�Ȳ�һ���� 1,
������Ӧ���ʵ� Qa �յ��� 1,��õ�صĵ綯��Ҳ���ڸ��¶��µ� E��ֵ֮��
8��ͬһ��Ӧ�� Cu2��Zn��Cu��Zn2, ��ѧ��Ӧ����ЧӦ�͵绯ѧ��Ӧ����ЧӦ�Ƿ���ͬ? Ϊʲô? ��: ����ͬһ��Ӧ����ѧ��Ӧ�ĵ�ѹ��ЧӦ���ط�Ӧ�ĵ绯ѧ��ЧӦ����ͬ����Ϊ��ѧ��ЧӦ��
���µ�ѹ�£��������ʱ�ķ�Ӧ�ȣ�Qp����H������ط�Ӧ��ЧӦ QR��T��S����H����nFE��QR ����֮��������뻷���ĵ��ܴ��ݵ� nEF��
��������9�� 2H��2e�� H2 �� Cl2��2e�� 2Cl ����������Һ�缫��Ӧ,�������Ʋ��(g,sln)��α�ʾ�� ��: ������Һ��Ӧ
����(g,Sln)����(g)����(Sln)��[��M��(Sln)��n��e(M)����M(g)]/nF
��[����M��(Sln)��n��n(M)����M(g)]/nF��RTlnaM/nF
���������缫,���Զ��Խ��� M(Pt)�缫Ϊ����,���Ӹ��ڽ��� M�ϣ�����ʽ
����Ϊ n��e(M) ������ n��e(g)��
10��������������绹�Ǵ��������ʲô������������������Щ�����йأ����ڽ�Ũ������ͭ
��Һ����ͭ�缫ʱ���������ʲô�磿
��: ������������磬����������������Һ�еĻ�ѧ��,�Ƚ��������ϵ��Ƹߣ�������(M,Sln)Ϊ��ֵ��
������������硣��缫�ı��Ԧ�����(M,Sln)�����μӵ缫��Ӧ�ĸ����ʵĻ���йء�Cu ���� ��Ũ������ͭ��Һ�У�������������硣
11��? (s)��? (M) ��ʱʲô���ƣ� ��? �� ? �ֱ�ʾʲô���ƣ���? �� ? �����кβ�ͬ��
����� ��? ���ɵ� ?��
��: ? (S)�� ? (Sln)��? (Sln) ��ʾ��������Һ�еĵ��ƣ�? (M) ��ʾ�ڽ��������ϵĵ��ƣ�
��? ��ʾ������������ͬ������ϵĵ��Ʋ?��ʾ������һ���ϵĵ���ֵ����?��ʾ
������ij���ϵĵ��������ֵ���涨����缫��ֵΪ�㣬��ij�缫�����缫��ɵ�أ� �����綯�ƣ���ô�õ綯����ֵ��Ϊ�õ缫��?ֵ��?ֵ��
12�����缫���Ʊ��и���������һ��ָ 298K�����ڲ�ͬ�¶��µı��缫����?���ܷ�ӵ缫
���Ƶ���˹�ع�ʽ���㣿
��: ���ԣ�ֻҪ֪���缫���Ƶ��¶�ϵ�� (? ?/?T)p������:
? (T)��? (298)��(??/?T)��T��?��(298)��(RT/nF)lnaM����(??/?T)��T
13�������ƺ��ƹ���������Ӧ�ĵ缫���ƹ�ʽ��α�ʾ����������Ӧ��?��?���Ƿ���ͬ��
�� ��: Na �缫�ķ�ӦΪ Na��e ���� Na(S)
?Na��/Na��?��Na��/Na��(RT/F)?ln(aNa��/aNa)
�� aNa����1�� aNa��1�� ?��?��Na��/Na
�����ƹ���缫����ӦΪ Na����e ���� Na(Hg)
?Na��/Na(Hg)��?��Na��/Na(Hg)��(RT/F)?ln(aNa��/aNa(Hg))
aNa(Hg) �� Na �ڹ����еĻ�ȡ�
�� aNa����1��aNa(Hg)��1�� ?Na��/Na(Hg)��?��Na��/Na(Hg)
����������缫?�� ����ͬ��? Ҳ��ͬ�� ��� Na �� Hg ���ܽ�ﵽ����ʱ,
��?��Na��/Na ��?��Na��/Na(Hg) ��ֵ��ȡ�
14��Ϊʲô�����ε缫���Ƕ������ӿ���ĵ缫�� ����
��: ��������Ϊ���࣬��ԭ��Ľ�����Ϊ����,��δ��ԭ�������ӽ�����Һ���������ε缫��
�Ƕ������ӿ���ĵ缫��
��������15����������ԭ�缫�� Pt��Fe2,Fe3�� Pt��Sn2,Sn4 ����Pt��ΪFe��Sn�Ƿ���ԣ�Ϊʲô��
����������: �����缫 Pt��Fe2,Fe3��Pt��Sn2,Sn4�е�Pt�ֱ�Fe��Sn����Fe�������������Ӧ ��
��Fe3��3e ���� Fe ?����0.036 V
�� Fe2��2e ���� Fe ?������0.447 V
���� Fe3��e ���� Fe2 ?����0.770 V
���������� Fe2����Fe �ĵ�����ͣ��ʸ�������ܵĵ缫��ӦΪ Fe����Fe2��2e
���������� Fe2����Fe3��e��
���� Sn �缫�Ķ�����ӦΪ��
�� Sn2��2e ���� Sn ?������0.1364 V
�� Sn4����2e ���� Sn2 ?����0.151 V
��˸�����Ӧ������ǵ�һ�������Ǻ��ߡ�
��������֧�缫��ʧȥ��ԭ������������ԭ�缫�����á����,���ܻ���
16�����е��������һ�����͵ĵ�أ�
(1) Ag, AgCl��HCl(m1)��H2(p��),(Pt)����(Pt),H2(p��)��HCl(m2)��AgCl,Ag(s)
(2) Hg��Zn(a1)��ZnSO4(a)��Zn(a2)��Hg �� a1��a2
(3) Na(����)(0.206��)��NaI(�� C2H5OH ��)��Na(s)
��: (1) ˫��Ũ���أ����ڵ����Ũ���ء�
(2) �缫Ũ���ء� (3) �缫Ũ���ء�
17��Ϊʲô˵�����缫Ҳ��һ������ѡ���Ե缫��ʹ�ò����缫Ӧע��ʲô���⣿�ò����缫
��Ϊָʾ�缫����ɵ���ܷ�ⶨ HCl �Ļ��ϵ����
��: ��Ϊ�����缫������죬�����ڲ����ڲαȵ缫����˲����缫���ƴ�С�������Һ��
��ȹ�ϵ��298KʱΪ: ? (����)��?��(����)��0.05915pH������?��(����)�dz���,��ֵ��С�� �缫����Ĥ�����й�, �� H�� Ũ����, �ɼ�������? (����)��С�������Һ�� H�� Ũ�� �й�, �������������ѡ��缫��Ҳ��һ������ѡ��缫��ʹ�ò����缫, Ҫע������ܽ� ǿ�Ļ�е��, ��Ҫ�Ѳ���Ĥ��Ӳ���ഥ������IJ����缫ʹ��ǰ, Ҫ������ˮ���� 24 С ʱ����, �����缫��ǿ����ʹ��, Ҫ�������, �Ϻ�����������ˮ��ϴ�� �ò����缫��ָʾ �缫, ��ɵ��, ���ܲⶨ��C����Һ�Ļ��ϵ������Ϊ�����ӵĻ���Dz��ܲⶨ�ģ���ϡ�� Һ��pH�IJⶨ�����Ǹ���һ���������壬Ҳ�����Թ�����ͳһ�涨�����������Һ��pHֵ Ϊ�ο��㣬ȷ��������Һ��p��ֵ���� pH(x)��pH(s)��(Es��Ex)F/2.303RT , ��Ϊ��������
��Һ��, pH(s) �������õ��� ��Lg��S(H��), ͬ��, pH(x) Ҳ�������õ��� ��Lg��(H), �����ò�
���缫����� HCl�� pHֵ���� ��Lg��(H��)�������ֵ�������Դ˼���� HCl �Ļ��ϵ�� ��
18������ͼ���� m��0 ʱ���� E�� ֵ,��ǰ�������ĸ����ʵĻ��Ϊ 1 ʱ���� E��E���ĸ���
�Ƿ�ì�ܣ� ͼ 9��10 �� E��0.1183lg m �ԡ�m ��ͼӦΪֱ�ߣ� ��ʵ�ʲ���Ϊֱ�� ��Ϊʲô�� �ڴ���������ȷ�ز�� E����
��: ���߸����ì�ܣ���ͼ������� E�� ֵ��ʵ�鷽����ѡ a��1 ʱ�� E��E�����Ƕ� E�� ����
����Ľ���,����ʵ���ֶβ��������� a�� 1 ����Һ�� �� E��0.1183lgm �� ��m ��ͼ���� m �� ��ʱ����ֱ�ߣ������Ƶ���ʽʱ���õ°ݣ����ȸ���ļ����ɣ��ö���ֻ������ϡ��Һ������� ����ֱ�ߣ�����ݣ٣�E��0.1183lg(m/m��)��f(m)������� Y �� m �ĸߴη�����ͼ��ʹ��������� ����������ϡ��Һ���ɵõ�ֱ�ߣ��Ӷ��õ�ԭʼ������ֵ֮��
19������ Fe��H2O ���ơ�pH ͼ�ش��������⣺
(1) ����ʲô���������ȶ�?
(2) ����ʲô�����и�ʴ������?
�� (3) ����ʲô���ơ�pH �����±���ʴΪ Fe2?
��: (1) Fe ��?����0.5, pHֵ �Ƚϵ�ʱ���ȶ���
(2) Fe �� pH �� 6����? ����0.4 ʱ�Լ�?��0����0.8, �� pH��8��14 �丯ʴ�����ء�
�� (3) �� pH������ ��0.4��?�� 0.8 ����Ϊ Fe2��
20����֪���б��缫��������
0.54 1.20 1.70
������ �� ����������3���������ɣ�3����������5�ɣ�6
1.23 1.69
���� M��2��������M���2��������M���4
���ñ��缫���Ƶ����ݣ��ش���������
(1) ����������μ����������Һ�У��ɵõ�ʲô���
(2) ���⻯����Һ��μ������������Һ�еõ�ʲô���
��������: (1) MnO4 �μ��� HI ��Һ�У��õ����� I3 �� Mn2��
������ ������: �����������ʱ, MnO4������, I��������, ������ IO3,
������������ ? (IO3/I3)��1.20�� ? (I3/I)��0.54 ����Ҫ������Ӧ: I��IO3 ���� 2I3
���� ������� H5IO6,�����λ����,�������� I ������Ӧ������� I ������
�������� ����,����ֻ���� I3����һ���� MnO4 ��ԭ����Ϊ Mn2��������� Mn2
���� �� MnO2,�� MnO2 ����� I ��Ҫ������Ӧ��? (MnO2/Mn2)��1.23 V
������? (I3/I)��0.54 ������ղ���ֻ���� Mn2��
���������� ���ԣ���ӦʽΪ: 15I��2MnO4��16H ���� 5I3��2Mn2��8H2O
������(2) HI �μ��� MnO4 �У�����Ϊ IO3 �� MnO2��
�������� �����ǣ����� MnO4 �ǹ����ģ� ��������� Mn2���� Mn2 �� MnO4 ��Ҫ����
�������� ��Ӧ���������Ϊ MnO2�� I ����������������� I3����? (IO3/I3)��1.20
���� ��? (MnO4/MnO2)��1.69V ҪС����Ҫ���������������� IO3��
������ ��? (H5IO6/IO3)��1.70 V����? (MnO4/MnO2) Ҫ����� I Ҳ���ᱻ������
H5IO6�� ��˷�ӦΪ:
��������I��2MnO4��2H���� IO3��2MnO2��H2O
��ʮ�� ������缫����
1. ����ΪӰ���ɢ���ȵ���������Щ��
�𣺷�ɢ���ȣ���Ҫ�ܵ缫�������ܶȺ͵������ҺŨ�ȵ�Ӱ�졣���������� �ܶ�Խ����Һ������Ũ��Խ��ɢ����ԽС���������Ժ��ԣ�ֻʣ�½��� �㡣�෴������������ĵ���ܶȺ���Һ������Ũ��ԽС����ɢ����Խ��
2. ����������������ɣ����ڽ�������Һ�����ϵĵ��ƽ���ηֲ��������֡����� ������ʱ�������ϵĵ����ֽ���ηֲ���
�𣺳�����ͼ10��6���������·�ת������ (1)������� (2)��������
3.����Ǧ��������Ϊ����˵��������Ϊ��Դ�ŵ�͵�س��ʱ���ĸ��缫�������� ����������������
��Ǧ��������Ϊ ��pb��pbSO4��H2SO4��pbO2��pb ������Ϊԭ��طŵ�ʱ��
�� ���� pb��SO42����pbSO4��2e ������������
���� ���� pbO2��4H��SO42��2e����2pbSO4 ������ԭΪ����
�����ʱ����Ϊ���أ�����������Ӧ��תΪ��ԭ���̣��ʳ�Ϊ�������������� Ӧ��תΪ�������̣��ʳ�Ϊ������
4. �������������У����ܷ���ɳ�һЩ���ԵĹ��ɣ�
�𣺲�����ԭ��ػ��ǵ��أ����Ƿ����������̵ĵ缫��Ϊ��������������ԭ���� �ĵ缫��Ϊ������
5. �ڲⶨ��������ʱ��ΪʲôҪʹ����һ�ο��缫���Բο��缫Ӧ����ʲôҪ�� ����Ϊʹ����һ�ο��缫��Ӧ�ö��������ɲ������缫������ͨ��ʱ�ĵ���ֵ�� �ⶨ�������Ȳ������缫����ͨ��ʱ��ƽ����ƣ��ٲ������缫�е���ͨ��
(�븨���缫��ɻ�·) �ķ�ƽ�����ֵ����������֮���õ�ij�����ܶ� iʱ �����ơ� �ο��缫Ҫ���λ�ȶ�����ֵ��֪�������Դ�i0�ϴ����籥�ʹ��缫��
6. ������⽻������i0���������岢˵������ֵ�Ĵ�С�ͼ����Ĺ�ϵ��
�𣺽�������i0�����ڵ缫��������Һ�е�ij����������ƽ��������ʱ���������뻹ԭ
�� ��Ӧ��Mn��ne����M ������������£���λʱ�䣬��λ�������������ĵ�����
���i0����ͨ���ܴ���������缫���Ƹı�С���õ缫��Ӧ�����Դ����� ������������֮��i0С���缫��Ӧ������С���õ缫��������������

7. ��ͨ�������ĵ����ܶȺ�Сʱ������(10��20)��(10��21)��(10��22)ʽ����(10��25)�� ��˵��Ӱ��U��ֵ�����ء�
��(10��20)ʽΪ ��i0exp(?F��?/RT)
(10��21)ʽΪ ��i0exp(����F��?/RT)
��i��Сʱ�������λ��?Ҳ��С����(10��20)ʽ��(10��21)ʽ������չ����
�� i��i0exp(?F��?/RT)��i0[1��?F��?/RT����(?F��?/RT)2??)��i0(1��?F��?/RT)
�� ����(10��21)ʽ ��i0exp(����F��?/RT)��i0(1����F��?/RT����(��F��?/RT)2??)
��i0(1���� F��?/RT) ����ʽ�ɺ�ȥ���η����ϵ��࣬�����(10��22)ʽ���� i�� �� ��i0(1��?F��?/RT)��i0(1����F��?/RT)
��i0(?F��?/RT����F��?/RT)��i0F(?����)��?/RT��i0F��?/RT ����?���£�1 ���� ��?��(RT/i0F)i����RT/i0F���� ��(10��25)ʽ�ǣ���i
�Ƚ�(10��21)ʽ��(10��20)ʽ��֪������Uֵ��Ӱ���������Ҫ��?��� �� ��?��������λ����
��������8. �����Ǧ�����صĵ��Һ�к������������ʣ�Mn2��Au3��Cd2��Fe2����
ָ�������������к����ʣ�Ϊʲô��
�������� Au3 ���ж����ʡ�
�缫����?��(pbSO4/pb)����0.3588��?��(pbO2/pbSO4)��1.6913��
������ ?��(Fe2/Fe)����0.447��?��(Cd2/Cd)����0.407��?��(Mn2/Mn)����1.1029��
���� ?��(Au3/Au)��1.507�� ������ʱ��������Ӧ��Au3��3e��Au�� ( ��Au ��λ��
�ڣ�0.5588���� Fe��Cd��Mn ��λС�� ��0.5585V )
������ ������Ӧ�� pbSO4��2H2O��2e����pbO2��4H��SO42 �� �� Au3���к��ġ�
9. ��˵����������ɢ���ʵIJ��졣
�𣺶���������ָ��Ӧ��������������Һ����������ƶ��Ĺ��̣�������Һ������ �Ӽ�����˶�����ɢ�����ӴӸ�Ũ�����Ũ�ȷ���ת�Ƶ�������������ɢ ��ָ����������ܼ����˶�������ָ����Һ��(�����ܼ�������)����˶���
10. �����۵������У��������̵Ĵ��ڶԵ缫��Ӧ���ʿ��ܵ�Ӱ�졣
�����ڶ����Ĵ��ڣ����Լ�����ɢ��ĺ�Ȧģ��Ӷ�Ҳ�������˵缫�����Ũ�� �ݶȣ�Ҳ���������ڵ缫���渽������ɢ�ٶȣ������������˵����ܶȣ����� �˵缫��Ӧ�����ʡ�
11. �ԱȽϵ绯ѧ������Ũ����Ļ���������
�𣺵绯ѧ���������ڵ缫������л�ѧ��Ӧ��绯ѧ��Ӧ��������̶�����ģ��绯
ѧ�����Ĵ�С�����ڵ缫���淴Ӧ�Ļ�ܣ������ܴ�С����缫�ϵ� ��� ��ѹ���ı䡣Ũ��������ڲμӵ缫��Ӧ�������ڽ������Һ���������Ũ�Ȳ� ��������ɢ���̶�����ļ�������ǿ���裬�ɼ�С�ģ��� i������Ũ�����
12. ��������תԲ�̵缫���ص���ŵ㡣
����ת�̵缫�ǽ�������缫�����һ��ʹ�缫���������������ת���Ӷ� ���ϸ������ɢ���Ⱦ��ȣ��̵缫�ϸ����Һ�ഫ���ٶ���ȣ������� �ȷֲ����ȣ�������õ缫����ķ�ӦDZ����Ҳ������˵����ܶȣ�������Ũ ����ơ�
13. ��˵����ѧ��ʴ�͵绯ѧ��ʴ�IJ�ͬ������
�𣺻�ѧ��ʴ�����ڽ�������ֱ�ӷ�����ѧ��Ӧʹ�����������ܽ⣬�����������ӻ�� �ۻ�����绯ѧ��ʴ�������ǣ���Ҫ�����ڷǾ��ȵĽ��������ϣ��ڽ������湹 �����(�ֲ����)�������ľֲ������������绯ѧ��Ӧ��ʹ�����������ܽ⣬�� �����ȵ���ʽ����������
14. �ԱȽ������������������������ص����ȱ�㡣
�����������DZ���ʴ�Ľ�������ص�������ʹ֮����������Ӧ�����ڸý��������跨 �Ⱦ����ʵ����������������γ�һ�����ܵ�Ĥ�������ͼӴ��˽������������ij��� �ƣ��Ӷ������˽����ĸ�ʴ����������һ���Ϊ�ۻ����������������ڱ���ʴ�Ľ� ��������һ����Ƹ����Ľ����������ý������ڱ���������������ʴ��Ҳ���DZ��� ���Ľ�����Ϊԭ��ص������������Ͳ��ᷢ����ʴ��������������һ����������һ ���Դ�ĸ������ӵ��������Ľ��������ڵ���ӵ�����ֲ�ԭ����е����������� �ȣ������ֲ�����ϵĿ��˵���Ϊ�㣬Ҳ�ͷ�ֹ�˽����ĸ�ʴ��
15. �����ڸ�ʴ�Խ����У����벻ͬ���Ӽ������ĸ�ʴ���ƺ�ʴ������Ĺ�ϵ�� �𣺼��������ͻ�ʴ�����ܼ���������������?a���Ӷ�����ʴ����I����������
�ͻ�������ܼ��ٽ�����������?C���Ӷ�����ʴ����I���������ͻ����
�ȼ��������������ƣ��ּ����������ƣ��Ӷ�����ʴ����i��
16. �Է����绯ѧ��Ӧ��һ�����������Դ��Ӧ������
�𣺵绯ѧ��Ӧ�������뻹ԭ��Ӧ�IJ���ֱ�����������ͬ�ļ�������������� ���跨����Ϳɵõ��绯ѧ��Ӧ���������뻹ԭ���������������ԭ��Ӧ�Ľ� �����ֲ������ͬһ���ڣ������跨���Է�����ܵõ����ַ�Ӧ�IJ��
17. �ԱȽϺ���Ʒ������������Ե�������
�𣺺���Ʒ����ǿ��Ƶ��ƣ������Ӧ�ĵ��������ʱ�൱����һ�ֻ�ԭ�����ܵõ� һ�ֲ��������������ǿ��Ƶ��������Ӧ�ĵ��ơ����ʱ�൱�ڶ��ֻ�ԭ���� �õ��Ƕ��ֲ�������һ�𡣶������Ķۻ����߲ⶨ��ֻ���ú���Ʒ������
18. �ò����缫����� ZnCl2 ˮ��Һ���������Ͽ��ܷ�����Щ�缫��Ӧ������п���� ���缫�����������ֽ���ʲô��Ӧ��
�����������ڲ������ܷ��� Zn2 �Ļ�ԭ�� H�Ļ�ԭ����? (H/H2��ƽ��)��? (Zn2/Zn��ƽ��)��
�����ڲ����ϵĦ�(H2)��С��0.05��0.045V�����? (H2������)��? (Zn������)������ �� H2��������п����H2�Ħ�(H2)Ϊ0.520��1.235V������Zn �Ħǡ�0������� pb ���ϣ�Zn ����������Ϊ? (Zn)����0.7628��0����0.763���������������Ϊ
? (H2)��0��1.235����1.235V������ Zn ���������ƣ���� Zn ���� H2 ������
���� CuCl2 ���� ZnCl2��Cu ��? (ƽ��)��0.3402���� Cu�� H2 ���������Ʒֱ�Ϊ ? (Cu)��0.3402��0��0.3402V ? (H2)��0��1.235����1.235V ��ǰ����Ȼ���ں��ߣ�
������� pb ����������ͭ������Cu2��2e��Cu��
19. ��ijһ������ԭ��Ӧ�ڻ�ѧ����н���ʱ������˵��ѧ�ܵ�ת��Ч�ʿɲ������� ѧ�ڶ����ɵ����ƣ�����˵�ɲ��ܿ�ŵѭ�����涨�����ת��Ч�������ơ����� ������������˵����
�𣺻�ѧ���������ת��Ҳ�ܵ��ȶ��������ơ���ѧ����У���ѧ�ܵ�ת��Ч�ʣ��� ���ص�������ù�Ϊ ��Gm����nEF�������ٴ��ڴ�ֵ���������Ƴ��ڶ����� �����������Ȼ������ѧ�ڶ����ɵ����ƣ���������������ת������������ת�� Ϊ���ĽΣ����ɻ�ѧ��ֱ��ת��Ϊ���ܣ������Dz��ܿ�ŵ�Ȼ�Ч�ʵ����ƣ��� ����ȼ�ϵ�ص���Ƴɹ����������ȼ�ϵ�ʹ�ü�ֵ��
20. �Ծ�Ӱ���Ȼ�Ч�ʺ�ȼ�ϵ�ص�����Ч�ʵ����ؽ��бȽϡ�
���Ȼ�Ч��Ϊ�ǣ�(T1��T2)/T2������Դ����Դ���²�Խ����Դ�¶�Խ�ͣ��Ȼ�
Ч�ʾ�Խ�ߡ���ȼ�ϵ�ص�Ч��Ϊ����Gm/��Hm��1��T��Sm/��Hm�������Sm/��Hm Ϊ��ֵ���ҵ�صIJ����¶�Խ�ͣ���Ч��Խ�������Sm/��HmΪ��ֵ����ǿ��� ����1�������������ǵ���ڹ���ʱ���ܴӻ��������ȶ�ת��Ϊ���ܡ�
��ʮһ�� �� �� �� ��
1�� �����ܡ����������ܡ��ȱ��������ܡ����������Ƿ���һ�������ͬ��
���ܵ�˵�����߶�����������Ĺ�ʣ�������������������𣬱�����Ϊ���ʱ������ �ڲ���������������� T��p �㶨ʱ���ⲿ��������Ϊ���������ܣ����漪��˹����
�� �ܣ�������T��p�㶨ʱ����λ����������ܣ����Ϊ�ȱ��������ܣ��䵥λΪ J?m2��
������ J��N?m���� J?m2 Ҳ�ɻ�Ϊ N?m1������������������ת��Ϊ��һ
�����Ϊ��ֱ�����ڵ�λ������������������������淽���������Ϊ������ ������Ȼ�ȱ��������ܺͱ�����������ֵ��ͬ��Ҳ�ɻ��ã��������в��ǰ���DZ� ����������ʸ����
2������������ֻ������ˮ��һ����Ȼ����������ֹ�����Զ��ֲ㣬����Ϊʲô�� ������ˮ�ǻ������ܵģ������߾�����ʱ���������ɢ��СҺ�Σ�����һ������ ����������ʱ��û���ܽ��ͱ����ܵĵ��������ʴ��ڣ������ʱΪ���ȶ���ϵ�� ��ϵ���Զ��������������ֲ����ϵ�������ͣ���˻��Զ��ֲ㡣
3��Ǧ�����ص������缫��һ���ǻ���Ǧ�缫����һ���ǻ�������Ǧ�缫������������ ���������⡰���ԡ����֣�
�����������ָǦ������Ǧ���ڶ���ԣ������д�ıȱ���������нϸ߱ȱ�������
�ܣ����ڻ�ѧ����״̬���������Ʊ��缫ʱ��������������γɵĸ߷�ɢ״̬�� ��������ѧ���ۼ��������ʣ���Ǧ���س���ʹ�û��߳��ڷ��ö�δ�ܼ�ʱ��磬 �缫�ĸ߷�ɢ״̬�����ͣ����ֻ���Ҳ����ʧ��
4 �ڻ��������У�����ԭ�ϵı��գ�Ŀǰ�ܶ���÷��ڱ��գ���������������������Щ�ŵ㣿 �𣺷��ڱ����ǽ�����ԭ�����С������ͨ��Ԥ�ȵĿ������������壬ʹ¯�ڹ�������� ������������״����ڣ������������˹�����ĽӴ����棬��ǿ�˴����봫�ȣ�ʹ�� ϵ���ڽϸߵĻ�ѧ����״̬��
5. �ڵι��ڵ�Һ��Ϊʲô���������ͷ��ѹʱҺ����ܵγ����������Σ�
�����ڵι��¶˵�Һ��ʰ��Σ���Һ��ĸ����������ϵģ�Һ�岻�״ӵιܵγ������ ��ҪʹҺ�δӹܶ˵��£�����������ͷ����ѹ����ʹ��ѹ�����ڸ���ѹ������ѹ�� ͨ��Һ�����������¶�Һ����������α���ĸ���ѹ����ʹ���α������α��棬 ����ʹҺ�ε��£��յ��µ�һ˲�䣬Һ�β������Σ��϶˳ʼ��Σ���ʱҺ�����λ�� ���ʰ뾶����һ������ͬ��λ������������������ѹ��Ҳ��ͬ�����ֲ�ƽ���ѹ���� ��ʹҺ���Զ����������Σ���������ʹҺ�ξ�����С�ı������
6. �ڽ�������ʵ��ʱҪ��������ƿ�м�Щ���Ƭ���ʯ�Է�ֹ������������ڣ�
���������Ƭ���ʯ������Һ���ڲ������γ�����( ���� )�������γ������ɲ�Ǽ䣬 ����������İ��α�������ʺܴ�����ݿ����Ĺ�ʽ������С�����ݱ��Է���ʧ�� �����ϵ�㲻������������·��ڣ���������¶��γɾֲ����ȵĽ��ȶ�״̬������ ���С������Һ���ڼ��Ϸ�ʯ�����ڷ�ʯ����ļ���нϴ���ˣ��˴� pr( ���� ��ѹǿ)����p*(Һ�履������ѹ)����������ڣ����ҷ�ʯ�ڲ������Ŀ�����Ҳ������ ���Ѹ�����Ϊ�γ��������ˣ������ʯ��ƿ������ӣ�����Ϊ�ֲ����ȴ��������� �Ӵ���ҺĤ��˲����ȣ�pr��p*�����Ϊ�ݣ�ʹ��ʯ������������Ϊһ������������
7�������һ�����м�С���Ǿ��������DZ�����Һ�У�Ͷ��һ��ϴ�����Ǿ��壬�ں� ���ܱյ������£�����һ��ʱ�䣬��ʱ�Ɑ��Һ��ʲô�仯��
���κ����ʵı�����Һ�������д����Ŵ�С��ͬ�ı��ܽ����ʾ�̬����ʱ��ʵ������ Щ��С��ͬ��ͬ�־�̬���ʵ��ܽ���Dz�ͬ�ģ��ɹ�ʽ��RT��n(Cr/C*)��2Mr��(S)/r��(S)
��֪��rԽС��������ԽСԽ�����ܽ��CrԽ�����ܽ�ȴ��ڴ�龧����ܽ��C*�� ��ˣ����ⱥ����Һ���ڷ��ú�����С�� �������ʧ������龧��ȴ������
8������������������Ũ�ȡ������ʣ���Ƿ���һ�����
�𣺱���������������Ũ�ȡ������ʣ����������ͬ�ĸ������Ũ���DZ�����ÿ��λ ������������ʵ�Ħ����������������������ʣ����ͬһ����������������� ָ��Һ��λ����������Һ�ڲ���Ӧ���ֵ��������ʵĹ�ʣ����������Ũ���뱾��Ũ ��֮������ɱ�����Լ��γɵ���Һ����������Ũ�������Ũ����ȣ���ֵ��С�� ��ˣ�����Ũ��ֵ֮�����������ֵ֮�����һ�¡�
9��������Լ�����Һ���Dz�ȡ����������������Һ�����ϣ������Խ�������ʽ�������� Һ�У�Ϊʲô��
�𣺱�����Լ�����Һ�����γɶ������У��Խ���ˮ�ı���������ʹ��ϵ�����ȶ����� ����Һ�ڲ���������Լ�Ҳ�ܰ���ˮ���ſ���һ���γɽ�����һ����˵��Ũ�Ƚ�ϡ ʱ�Զ������������ڱ���Ϊ��������Һ�ڲ�Ҳ�����мĽ����γɣ����ű������ ����Ũ�����ӣ����涨���������ӵ�ͬʱ���ڲ���������Ҳ���࣬һ�������γɵ��� ��Ĥ����ʱ��ﵽ�ٽ罺��Ũ�ȣ��Ժ������ӱ�����Լ�Ũ�ȣ�ֻ���ӽ����������� ���������ٸı䡣
10��������������������ͱ�����Լ��кβ�ͬ��
�����������ͱ�����Լ���ijһ���� pH ֵʱ������Һ�гʵȵ�㣬����Һ������� pH ֵʱ������������Һ�У����������ӱ�����Լ������ڴ��ڸ� pH ֵ����Һ�У��� �������ӱ�����Լ��� �������ͱ�����Լ�����ҹ�в��ܵ��룬���ܳ�Ϊ������ ������Լ����������ͱ�����Լ��ຬ����ˮ�в�������ǻ�(��OH)�ͣ���O��)��� �ĺ������ţ������������Ϊ��ˮ���š�
11��ˮ�ڲ������гʰ���ҹ�棬��ˮ������Ρ�Ϊʲô��
����ˮ�벣���ĽӴ�С��90�㣬ˮ�ı���ĸ���ѹ��Ϊ��ֵ��������ʹˮ�ڲ�����
�гʰ��档��ˮ���벣���ĽӴ�Ϊ����90�棬ˮ������ĸ�����Ϊ��ֵ�����ˮ���ڲ� �����ڳ���Һ�档
12����װ�в���Һ���ëϸ���У���һ�˼���ʱ����ͼ 11��8 ��ʾ��(a) ��ʪ��Һ���� ëϸ����һ���ƶ��� (b) ����ʪҺ������һ���ƶ��� Ϊʲô��
(a) (b)
��(��)�������ˮҺ������Զ����ȵ�(������)�ƶ���(��)�������ˮ���������ż��� ��(������)�ƶ�����˱����������¶ȵ����߶����͡����ȴ������������٣�δ���� �����䡣�ڱ������������£�(��)��ʪ��Һ�������ƶ���(��)����ʪ��Һ�������ƶ���
13������ˮ������棬���ʵ�������Լ�(���ˮ��)������Ϊʲô���Ըı�������� �ʣ�ʹ�������ˮ�ԣ�
�����ڱ�����Լ�����ˮ�����ϵĶ������У���ˮ������������Ӵ�����ˮ������ ��������ʹ��������γ�һ����ˮ�㣬�Ӷ��ı��˹������ṹ�����ʣ�ʹ����� ��ˮ�ԡ�
14�����ڱ�����Լ��Ļ������ʣ�Ϊ������ά���������ʵ�������Լ������ɴ�ͼ��� ����ʹ��ά����ƽ����
��������ά���������ˮ�ԣ�������Լ�����ˮ������������ά������������У����� �DZ�����Լ�����ˮ���Ŷ������У�ʹ��ά�ı����γ�һ���µ���ˮ���棬����ˮ�� ��һ���ǽ���ֱ����֬�����ţ�����������ά�����γ�һ�㱡�����Ͳ㣬���Գ����� ��ƽ�����ָС�
��ʮ���� ���弰�������Һ
1����Һ������ɢ���? Ϊʲô? һ����Һ���? �ܽ��������������ϵ����Ϊ�Σ� ��(1) ���ڴ�Һ������壬��n1��n2����Ӧ��ɢ������ʵ��������Ҳ���������� �⣬���������ܶȵ�������ʹ�������ʱ仯������ġ�
(2) ��һ������Һ��ɢ����Ǻ����ģ�һ��ͷ�����Һ������������С���������� Զ�����ܽ����ԣ���˶����ЧӦ���б��ܽ�������Һ�ļ�����
(3) �ܽ������ϴ�ɢ����ǿ��������⣬���ܽ��Ǿ��ȷ�ɢ��ϵ������������
2��������Ϊ��Ҫ��ǿ�Ĺ�Դ��
����Ϊ���� Raylligh ��ʽ����һ����ϵ����ɢ����ǿ����������ǿ�ȳ����ȡ��� �����ǹ۲콺����ɢ��⣬���ʹ�ó�������ǿ��Դ������Ч����߳������� �۲�������
3��Ϊ��˵���ǹ۲쵽�IJ����˶������ǽ�����ʵ���˶����?
�𣺲����˶������ڷ�ɢ���ʵķ������˶����ϵ��ɸ�������ͬʱ��������������δ�� ��������ʱ����ijһ������һ�������ã������½����˶�����˲����˶����ǽ�����
�� ��ʵ�˶����������ܲ�ƽ��ij������������ԼΪ108�룬�����ǵ������ܷ�
����������Ϊ 0.1�룬����������۲쵽�Ľ���ƽ��λ���ǽ�����ÿ�������ܸ��� ������������˶��仯�ĺ�۽����
4�������˶���ɢ����ʽ�����Խ����Щ����?
�𣺸��� ����2Dt��RTt/3��r��L����������� ��
(1) ֤ʵԭ�ӡ� ���Ӵ��ڵ���ʵ�ԡ�
(2) �����˶�ѧ˵�Ŀɿ��ԡ�
(3) ���㰢��ӵ�������ʵ���ֶΡ�
(4) ���ø���ϵ���о���ɢ��ϵ�Ķ������ʣ�������ɢϵ���Լ����Ӵ�С�ȡ�
5��Ϊʲô����������(������)M��(4/3)?��r3��L ?
���� (4/3)��r3�� Ϊһ����������������L Ϊ����ӵ����������ݣӣ��ƣ�Ħ��
�ĸ�����ָ��ϵ�����������ʵĻ�����Ԫ����Ŀ�� 0.012kg 12C ��ԭ����Ŀ(��)�� �ȣ���ν������Ԫ������ԭ�ӡ� ���ӡ� ���ӡ� ���Ӽ��������ӻ���Щ���ӵ��ض��� �ϣ���˶��ڽ�����ϵ��˵������6.022?1023(����)��Ŀ�Ľ��������ν�� 1 mol �Ľ���������(4/3)��r3�ѣ� ���ǽ���������(������)��
6���ԱȽϽ���������ƣ�Stern��(���ܲ�)���Ƽ��ĵ���?
�𣺽������������ָ����������ڵ�����������ӵ�ԭ��ʹ�ܽ����Ӵ��磬��������� ��Һ��ĵ��Ʋ������ѧ��ƽ����ƲStern �������ָ�ܽ����ӱ������ھ���

����Ӽ������ö���������һ����ܵ��ƣ���Ȼ������С�ڽ���������ơ�
�ĵ���(���������)��ָ���ܽ�������Һ�����˶�ʱ�����������ܲ�֮����һ�㻬���棬
�Ի�����ĵ�����Һ���ڲ�����Ϊ�㴦�ĵ��Ʋ��Ϊ��������ƻ�Ʀ�(zeta)���ơ�
7�������軯ͭ�ܽ����ȶ����������軯�أ���д���佺�ű�ʾʽ��������ɷ������?
������K4Fe(CN)6Ϊ�����軯ͭ���ȶ��������Ȼ��Fe(CN)64�����ܽ����ӵĽ��ܲ��ڣ�
�������� ����Ϊ{[Cu2Fe(CN)6]m?nFe(CN)64?(4n��q)K}q?qK ������������ɡ�
8����������ܽ��Ƕ���ѧ���ȶ�������ѧ�ϲ��ȶ�����ϵ�����о۳����ȶ�������? �������ܽ��IJ����˶��Լ���ɢ�˶��������ڽ��������˫���ṹ�������ܼ���Ĥ, ����ܽ��Ķ���ѧ�ȶ��ԣ����ܽ��Ǹ߶ȷ�ɢ�ķǾ�����ϵ�����кܴ�ı������� ��,���Է��۳��Խ�����ϵ���������ƣ����������ѧ�IJ��ȶ���ϵ���о۳����ȶ� �����ԡ�
9���������ձ��и�װ��20cm3ͬ���ܽ�������������Һʹ�俪ʼ�����۳�ʱ���ڵ�һ��
���� �ձ���� 1mol?dm3KCl��Һ 2.1cm3���ڶ������12.5cm3��0.005mol?dm3 Na2SO4
���� ��Һ���������ձ����7.5cm3��3.3?104mol?dm3Na3pO4��Һ����ȷ����۳�ֵ����
�ܽ���ɷ��š�
�����ܽ��м���������Һ�����ܽ���ʼ���Ծ۳�ʱ��ÿ���ܽ��м������ʵ���С�� ��Ũ�ȳ�Ϊ���ֵ���ʵľ۳�ֵ(CFC)��
���� 1mol?dm3KCl��Һ�۳�ֵ��1?2.1?1000/(20��2.1)��95.02m?mol?dm3
���� 0.005mol?dm3Na2SO4�۳�ֵ�� 0.005?12.5?1000/(20��12.5)��1.923m?mol?dm3
���� 3.3?104mol?dm3Na3pO4��Һ�۳�ֵ��
�� 0.00033?7.5?1000/(20��7.5)��0.091m?mol?dm3 ���ܽ��Ǵ�����ɵġ�
10����������ƽ�⽨���������Һ��ѹ��ϵ��Σ�
�𣺵�����ƽ�⽨�������ڵ���ʶԸ߷�����Һ��ѹ��Ӱ�죬��Һ�����Ũ�Ȳ�� �ڴ��߷�����Һ��Ũ�ȣ�����е����ʱ�������Һ����ѹһ���������ʵ������
11����Ĥ�ڡ� ��û�й�ͬ���ӣ���Ĥ��ֻ���ܼ�ˮʱ������ƽ��Ĺ�ϵ��Σ�
��(1) ��Ĥ������ͬ���ӣ�
���磺
Ĥ �� ��
�������� ��ʼ Na R K Cl
C1 C1 C C2
���������� ��ƽ�� Na R K Cl K Cl Na
c1��z c1 x y c2x c2��y z
����������ԣ��� x��(c1��z)��c1��y �� y��x��z
��������ƽ�� (c1��z)y��z(c2��y)
(c1��z)(x��z)��z[c2��(x��z)] (1)
�Լ� xy��(c2��x)(c2��y) x(x��z)��(c2��x)[c2��(x��z)] (2)
��(1)(2)�������� x��c2(c1��c2)/(c1��2c2)
y��c22/(c1��2c2)
z��c1c2/(c1��2c2)
(2) ��Ĥ��ֻ���ܼ�ˮ�磺
Ĥ �� ��
���� ��� Na R 2O
���� c1 c1 cHcOH��Kw
���������� Ĥƽ�� Na R H OH
c1��x c1 x x x
����ƽ�������� (c1��x)(Kw/x)��x2 ��[Kw(c1��x)]1/3
��x����c1 �� x��(Kwc1)1/3
�� ��Ĥ�����ԣ�Ĥ����ԣ�������ӵ����ΪR��Cl�ͣ������෴������Ĥƽ���Ϊ
Ĥˮ�⣬��˿��Բ��ֽ���������ϸ������ʲ�ͬ�ģ�H��θ����γɡ�