������ѧÿ���ܽ�
��1������ѧ��һ���ɼ�Ӧ��
1��ϵͳ������������
����ѧ�а��о��Ķ������ʺͿռ䣩��Ϊϵͳ����ϵͳ������ص��������ʺͿռ��Ϊ����������ϵͳ�뻷��֮���Ƿ����������������ʽ���ϵͳ��Ϊ���ࣺ����ϵͳ�����ϵͳ�ͳ���ϵͳ��


2������ѧƽ��̬
ϵͳ�ĸ��ֺ�����ʲ���ʱ����仯����Ƹ�ϵͳ��������ѧƽ��̬������ͬʱ�����ĸ�ƽ�⣺��ƽ�⡢��ƽ�⡢��ƽ�⡢��ѧƽ�⡣
3�����빦
(1) ���빦�Ķ���
�ȵĶ��壺����ϵͳ�뻷�����¶Ȳ�Ĵ��ڶ����������������ʽ����Q��ʾ��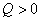 ��ʾ������ϵͳ���ȡ�
��ʾ������ϵͳ���ȡ�
���Ķ��壺����ϵͳ�뻷��֮��ѹ����Ĵ��ڻ�����������Ĵ������������������ʽ����W��ʾ��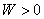 ��ʾ������ϵͳ������
��ʾ������ϵͳ������
(2) �������������
���ж�����ʽ��ͨ���漰�������������ϵͳ����仯ʱ�Ĺ����䶨��Ϊ�� 
ʽ��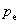 ��ʾ������ѹ����
��ʾ������ѹ����
���ڵ���ѹ���� 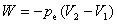
���ڿ�����̣���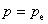 ��pΪϵͳ��ѹ��������
��pΪϵͳ��ѹ��������

��������������������繦�����湦�Ƚз����������W���ʾ��
4������ѧ��
����ѧ���Է���U��ʾ����ϵͳ��״̬��������ϵͳ��״̬1�仯��״̬2������̵�����ѧ����Ϊ 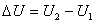
����һ������ϵͳ������ѧ����������������������״̬��������
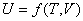
����ȫ��Ϊ
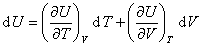
��һ�������������壬����
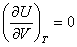 �� U=f��T��
�� U=f��T��
��һ������̬�������������ѧ��ֻ���¶ȵĵ�ֵ������
5������ѧ��һ���ɼ���ѧ����ʽ
(1) ����ѧ��һ���ɵľ�������
�� �������Դ�һ����ʽת��Ϊ��һ����ʽ������ת���ʹ��ݹ������������䡣
�� �����������������������������Ļ�����Ϊ��һ������������һ���������Dz����ܴ��ڵġ�
(2) ��ѧ����ʽ
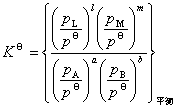
���ڷ��ϵͳ������ѧ��һ���ɵ���ѧ����ʽΪ
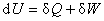 ��
�� 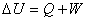
�����ϵͳ������ѧ�ܵĸı������ڹ����л�������ϵͳ���Ⱥ����ܺ͡�
6����
���Է���H��ʾ����ϵͳ��״̬����������Ϊ

����һ������ϵͳ������������������������״̬��������

����ȫ��Ϊ

��һ�������������壬����
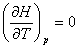 �� H=f��T��
�� H=f��T��
��һ������̬�����������ֻ���¶ȵĵ�ֵ������
7���������
��ͨ��ԭ�����̵ķ�����仯��ʹϵͳ�ָ���ԭ��״̬��ͬʱ������û�������κ������Ա仯�Ĺ��̣���Ϊ����ѧ������̣���֮����Ϊ��������̡�
�������������������
(1) �����������һ�������������ӽ�ƽ��̬�Ĺ��������ɣ������������������ġ�
(2) ֻҪ����ͬ����ѭ����ԭ��;������һ������̣���ʹϵͳ�ͻ���ͬʱ�ָ�ԭ״��
(3) �ڵ��¿�������У�ϵͳ�Ի���������Ϊ���������ϵͳ������Ϊ��С����
8������
(1) ���ݵĶ���
��û�з�������������£���һ�����������ڲ���������ѧ�仯������£�����dQ �����¶���T����T��dT����dQ��dT �ı�ֵ��Ϊ�����ʵ�����C��
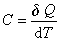
(2) ����Ħ������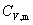
��n molij�����ڵ������������¶�T����T��dT�����յ���Ϊ dQV�������Ħ������Ϊ
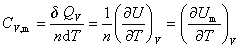
(3) ��ѹĦ������
��n molij�����ڵ�ѹ���������¶�T����T��dT�����յ���ΪdQp�����ѹĦ������Ϊ
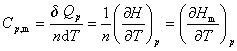
(4) ������������ϵ
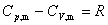
(5) �������¶ȹ�ϵ�ľ���ʽ
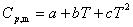 ��
�� 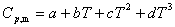
9������ѧ��һ������p��V��T�仯�е�Ӧ��
(1) ���ݹ���
W=0 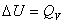
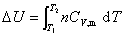 ���������������塢Һ�塢����ĵ��ݹ��̣��������������p��V��T�仯���̣�
���������������塢Һ�塢����ĵ��ݹ��̣��������������p��V��T�仯���̣�
(2) ��ѹ����
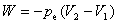
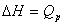
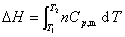 ���������������塢Һ�塢����ĵ��ݹ��̣�������������������������仯���̣�
���������������塢Һ�塢����ĵ��ݹ��̣�������������������������仯���̣�
(3) ��������ĵ��¹���
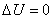
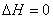
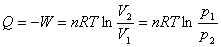
(4) ���ȿ������
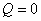
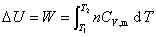
������������ľ��ȹ��̣����ܹ����Ƿ������ʽ�������á�
������������ľ��ȿ�������У�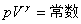 ��
��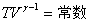 ��
�� ��
��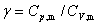
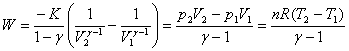
10������ѧ��һ��������仯�е�Ӧ��
(1) �����(��)
ϵͳ�����ۼ�̬�仯��Ϊ��仯�������������ۻ�������������ת���ȣ���仯���������ջ�ų����ȣ���Ϊ����ȣ��ʣ�
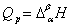
(2) ��仯���̵������������ѧ��
��ϵͳ�ڵ��µ�ѹ��������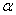 ��仯��
��仯��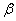 �࣬����仯�����������Ϊ
�࣬����仯�����������Ϊ
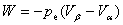
������Ϊ���࣬�����������������������Ϊ�������壬����
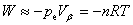
��仯�����е�����ѧ��Ϊ
�� 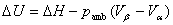
�����������������
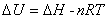 ]
]
11������ѧ��һ�����ڻ�ѧ�仯�е�Ӧ��
(1) ��Ӧ����
��nB ( )��nB������Ϊϵͳ����һ����B�ڷ�Ӧ��ʼʱ(��0)����Ӧ���е���ʱ���ʵ�������BΪB�Ļ�ѧ����������
)��nB������Ϊϵͳ����һ����B�ڷ�Ӧ��ʼʱ(��0)����Ӧ���е���ʱ���ʵ�������BΪB�Ļ�ѧ����������
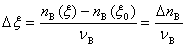
ʽ�У�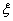 Ϊ��Ӧ���ȡ�
Ϊ��Ӧ���ȡ�
(2) ���ʵ�����ѧ��̬�Ĺ涨
����ı�̬�������Ǵ�����B�����������е���һ���B�������¶�ΪT��ѹ��p���²����ֳ������������Ե����崿����B�ģ����룩״̬��
Һ��(�����)�ı�̬�������Ǵ�Һ��B����Һ�������е����B�������¶�T��ѹ��p����Һ�壨����壩������B��״̬��
(3) ��Ħ����Ӧ�ȣ��ʣ��ļ���
�� ��Ħ�������ȣ��ʣ��Ķ���
���¶�T�ı�̬�£����ȶ���̬�ĵ�������1 mol����Ļ����� B ���ʱ��Ϊ������B(��)���¶�T�µı�Ħ�������ȣ��ʣ�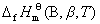 ��
��
�� ������
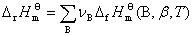
�� ��Ħ��ȼ���ȣ��ʣ��Ķ���
���¶�T�ı�̬�£���1mol �������B����������ȫ������ȼ�գ���Ӧ���ʱ䣬��Ϊ����B(��)���¶�Tʱ�ı�Ħ��ȼ���ȣ��ʣ�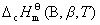 ��
��
�� ��������
�� 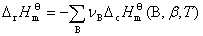
�� ��Ӧ�����¶ȵı仯��ϵ����ϣ������
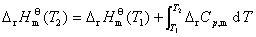
�� ��Ӧ�ı�Ħ���ʱ����Ħ������ѧ�ܱ�Ĺ�ϵ 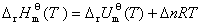
��2�� ����ѧ�ڶ�����
1������ѧ�ڶ����ɵľ������
������˹(Clausius)˵�����������ܰ��ȴӵ������崫����������������������仯����
������(Kelvin)˵�����������ܴӵ�һ��Դȡ����ʹ֮��ȫ��Ϊ���������������仯����
2���صĶ���
���Է���S��ʾ������ϵͳ��״̬�����������������Ϊ
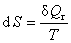
3������ѧ�ڶ����ɵ���ѧ����ʽ
���ϵͳ������ѧ�ڶ����ɵı���ʽΪ


ʽ��TΪ�������¶ȣ��Կ�����̲��õȺţ���Te=Tsys���Բ�������̲��ò��Ⱥš�
4������ԭ�������о�
(1) ����ԭ��
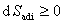 ��
�� 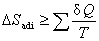
��ϵͳ�����ȹ�����ijһ״̬������һ״̬ʱ�������ز����٣����ھ��ȿ�������в��䣬�����Ȳ�������̺����ӣ����Ϊ����ԭ����
(2) ���о�
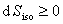 ��
�� 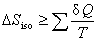
��ʽ��Ϊ���оݡ��京����
�� ʹ����ϵͳ�������κ�һ��С�仯ʱ���� ����ù���ϵͳ����ƽ��̬��
����ù���ϵͳ����ƽ��̬��
�� ���¹���ϵͳ������Ĺ����п����Զ�������
5��ϵͳ�ر�ļ���
����ϵͳ�ر�Ļ�����ʽ
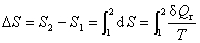
(1) p��V��T�仯�ر�ļ���
�� ���������p��V��T�仯
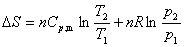
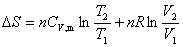
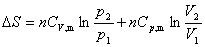
�� Һ������p��V��T�仯
��ѹ���¹��� 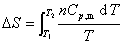
��Ϊ���������ֿɵ� 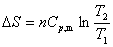
���ݱ��¹��� 
��Ϊ���������ֿɵ� 
(2) ��仯�����ر�ļ���
�� ����������
�� ������������
����ƿ���;�������㣬��

Ѱ�����;����ԭ����;���е�ÿһ��������棻����;���е�ÿ���� ��������Ӧ�Ĺ�ʽ�����ã���������Ӧ��ÿһ�������������ݡ�
��������Ӧ�Ĺ�ʽ�����ã���������Ӧ��ÿһ�������������ݡ�
6�������ر�ļ���

7������ѧ��������
(1) ����ѧ�������ɵľ�������
����ѧ�������ɿɱ���Ϊ����0Kʱ���κδ����ʵ������������ֵΪ�㡱��
(2) ��ѧ����ʽ
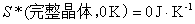
8���涨�غͱ���
��������ѧ�ڶ����ɣ�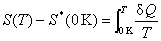 ���ָ�������ѧ��������
���ָ�������ѧ�������� �����������B�Ĺ涨�أ���
�����������B�Ĺ涨�أ���

1mol ����B���ڱ�״̬�µĹ涨�ؽ�����Ħ���أ�����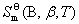 ���䵥λ��
���䵥λ��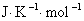 ��
��
��֪����B�� ��
��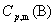 ����������¶�T������
����������¶�T������

9����ѧ��Ӧ���ر�ļ���
����������B�ı�Ħ�������ݼ��㻯ѧ��Ӧ���ر�
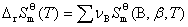
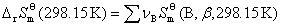
�� 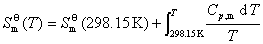
10����ķ���������ܺͼ���˹������
(1) ��ķ���������ܺͼ���˹������
��ķ���������ܶ��� 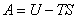
����˹�����ܵĶ��� 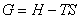
(2) ��ķ���������ܺͼ���˹�������о�
��ķ�����������о� ��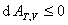 ��
�� 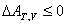
����˹�������оݣ�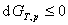 ��
�� 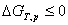
���ϸ�ʽ�ֱ�к�ķ�����������оݺͼ���˹�������оݡ�
11������ѧ������ϵ
(1) ����ѧ��������
����������������״̬��������ȷ��ϵͳ״̬��ϵͳ������ѧ��������Ϊ
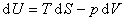
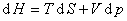
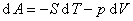
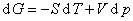
��ʽ�У��ֱ����dS=0��dV=0��dS=0��dp=0��dT=0��dV=0��dT=0��dp=0���������Եõ�
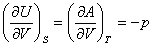
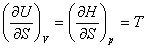


(2) ���˹Τ��ϵʽ

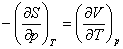
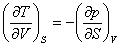
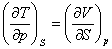
���ϸ�ʽ�����˹Τ��ϵʽ
(3) ����˹-��ķ���ȷ���

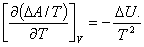
12������仯������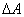 ��
��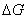 �ļ���
�ļ���
(1) ���µ�p,V�仯���������ļ���
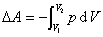
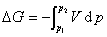
���϶�ʽ�����ڷ��ϵͳ�� �����塢Һ�塢����ĵ��±仯��
�����塢Һ�塢����ĵ��±仯��
�����������壬 �����϶�ʽ����
�����϶�ʽ����
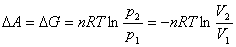
��A��G�Ķ���Ե��¹�����
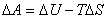
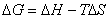
(2) ��仯�������ļ���
�� ���µ�ѹ�Ŀ������
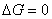
�� ���������
�Բ�������仯���̣�Ӧ��ƾ�����ͬʼ̬����̬�Ŀ�����������㦤G��
��3�������ϵͳ����ѧ
1��ƫĦ����
(1) ƫĦ�����Ķ���
�����ϵͳ�����1��2������k�ȹ��ɣ���ϵͳ��������������X(�磬V��U��H��S��A��G)Ӧ��T��p��n1��n2������nk �ĺ�������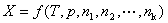 ����ȫ��Ϊ
����ȫ��Ϊ
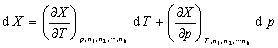
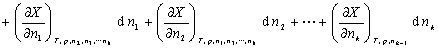
����  ΪB���ƫĦ������
ΪB���ƫĦ������
(2) ƫĦ�����ļ��Ϲ�ʽ
�ڵ��µ�ѹ�Ķ���ֵ���ϵͳ�У�ֻҪ֪������ֵ�ƫĦ����XB��m�����ʵ���nB�Ϳ�ȷ��ϵͳ����������X��ֵ
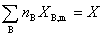
��ʽ��ΪƫĦ�����ļ��Ϲ�ʽ��
(3) ����˹���ź�ķ��ʽ
����ϵͳ�У���ͬ���1��2��������k��ͬһƫĦ����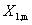 ��
��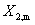 ��������
�������� �����¹�ϵ
�����¹�ϵ
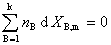 ��
�� 
��ʽ��nBΪ���B�����ʵ����� Ϊ���B�������������϶�ʽ��Ϊ��
Ϊ���B�������������϶�ʽ��Ϊ��
2����ѧ�Ƽ����о�
(1) ��ѧ�ƵĶ���
һ������ɲ���ľ���ϵͳ����������������V��U��H��S��A��G�ȶ��ɱ�ʾΪ�������������ĺ�������
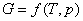
��ɿɱ�ľ���ϵͳ��G�ĸı�������¶Ⱥ�ѹ���й��⣬�������ֵ����ʵ����ĸı��йأ��� ������
������
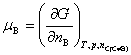
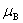 ��Ϊϵͳ��B��ֵĻ�ѧ�ơ�
��Ϊϵͳ��B��ֵĻ�ѧ�ơ�
(2) ���廯ѧ��
���廯ѧ�����£�
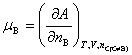
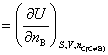
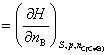
���ϸ�ʽҲΪ���B�Ļ�ѧ�ƵĶ���ʽ����Ϊ���廯ѧ�ơ�
(3) �����ʵĻ�ѧ��
�����ʵĻ�ѧ�ƾ�������Ħ������˹�����ܣ���
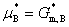
(4) ��ѧ���о�
�� ��ƽ������
��ϵͳ��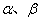 ������ƽ�⣬�����
������ƽ�⣬�����

��ʽ��Ϊ��ƽ��������������ƽ�⣬�����B�������л�ѧ����ȡ�
�� ��ѧƽ������
��ѧ��Ӧ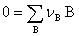 ƽ������Ϊ
ƽ������Ϊ
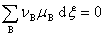 ��
�� 
��ʽ������ѧ��Ӧ���ƽ��ʱ�����뻯ѧ��Ӧ����ֻ�ѧ�ƴ����͵����㡣
(5) ��ѧ�����¶Ⱥ�ѹ���Ĺ�ϵ
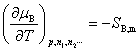
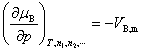
3������Ļ�ѧ�Ƽ��ݶ�
(1) ��̬��������Ļ�ѧ��
��̬����������һ���¶��µĻ�ѧ����ѹ���Ĺ�ϵΪ
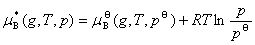
ʽ��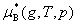 Ϊ��̬����������T��p�µĻ�ѧ�ƣ�
Ϊ��̬����������T��p�µĻ�ѧ�ƣ�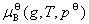 Ϊ��̬����������T��
Ϊ��̬����������T��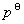 �µĻ�ѧ�ƣ�����̬��ѧ�ơ�
�µĻ�ѧ�ƣ�����̬��ѧ�ơ�
��2����̬��������������������ֵĻ�ѧ��
��������������һ���¶��������B�Ļ�ѧ�����ѹ�Ĺ�ϵΪ
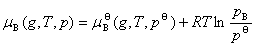
ʽ��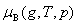 Ϊ�����������������B���¶�ΪT����ѹΪpB�µĻ�ѧ�ƣ�Ϊ�����������������B��T���µ��������־��������������ԵĵĻ�ѧ�ƣ�����̬��ѧ�ơ�
Ϊ�����������������B���¶�ΪT����ѹΪpB�µĻ�ѧ�ƣ�Ϊ�����������������B��T���µ��������־��������������ԵĵĻ�ѧ�ƣ�����̬��ѧ�ơ�
(3) ��̬��ʵ����Ļ�ѧ�����ݶ�
��̬������������һ���¶��µĻ�ѧ����ѹ���Ĺ�ϵΪ
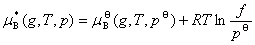
�� 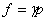
ʽ��f���ݶȣ�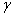 ��Ϊ�ݶ�ϵ����
��Ϊ�ݶ�ϵ����
(4)��ʵ����������������ֵĻ�ѧ��
�Է���������Ļ�������������B���仯ѧ�Ƶı���ʽΪ
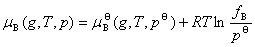
ʽ��fBΪ�������������������B�÷��ݶȡ�
4�����ڶ����ɺͺ�������
(1) ���ڶ�����
ƽ��ʱ��ϡ��Һ���ܼ�A�������еķ�ѹpA���ڴ��ܼ���ͬһ�¶��µı�������ѹ ������Һ���ܼ���������
������Һ���ܼ��������� ������ѧ����ʽΪ��
������ѧ����ʽΪ��
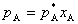
(2) ��������
��һ���¶��£�ϡ��Һ�лӷ�������B����������ƽ��ʱ�����еķ�ѹ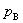 �����������Һ�е�����������Ũ�ȣ������� �����Ϊ�������ɡ� ����ѧ����ʽ�ֱ�Ϊ��
�����������Һ�е�����������Ũ�ȣ������� �����Ϊ�������ɡ� ����ѧ����ʽ�ֱ�Ϊ��
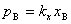
ʽ�� ��
�� ��
��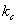 Ϊ�Բ�ͬ��ɱ�ȱ�ʾ�ĺ�����������λ�ֱ�ΪPa��Pa·kg·mol-1��Pa·mol·dm-3��
Ϊ�Բ�ͬ��ɱ�ȱ�ʾ�ĺ�����������λ�ֱ�ΪPa��Pa·kg·mol-1��Pa·mol·dm-3��
5��������Һ
(1) ������Һ�Ķ���
�������¶��£���Һ�е���һ���B��ȫ��Ũ�ȷ�Χ�ڶ��ϸ�������ڶ�����������Һ��Ϊ������Һ��
(2) ������Һ���������B�Ļ�ѧ��
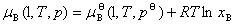
ʽ��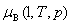 Ϊ������Һ���������B���¶�ΪT����ѹΪp�µĻ�ѧ�ƣ�Ϊ������Һ���������B����������
Ϊ������Һ���������B���¶�ΪT����ѹΪp�µĻ�ѧ�ƣ�Ϊ������Һ���������B����������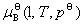 Ϊ��=1ʱ�������BΪ��Һ��״̬������T�������µĻ�ѧ�ƣ�����̬��ѧ�ơ�
Ϊ��=1ʱ�������BΪ��Һ��״̬������T�������µĻ�ѧ�ƣ�����̬��ѧ�ơ�
6��������Һ��ͨ��
�ڵ��µ�ѹ�����£���һ�����ϵĴ���ֻ���γ�������Һʱ����������ʲ��䣬������������˹�����ܼ��٣��˼�Ϊ������Һ�Ļ�����ʣ�������ѧʽ��ʾΪ
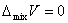
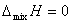

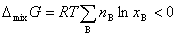
7������ϡ��Һ
(1) ����ϡ��Һ�Ķ���
һ���¶��£��ܼ������ʷֱ��������ڶ����ɺͺ������ɵ���Һ��Ϊ����ϡ��Һ�����ϡ��Һ��
(2) ����ϡ��Һ���ܼ������ʵĻ�ѧ��
�� ����ϡ��Һ���ܼ��Ļ�ѧ��
��Ϊ����ϡ��Һ���ܼ��������ڶ����ɣ������仯ѧ�Ƶı���ʽ��������Һ��������ֵĻ�ѧ����ͬ����

�� ����ϡ��Һ�����ʵĻ�ѧ��
��Ϊ����ϡ��Һ���������غ������ɣ����Դ�����Һƽ������������������ɿɵõ�ϡ��Һ�����ʵĻ�ѧ�ƣ���
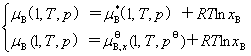
��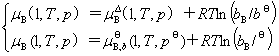
��
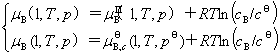
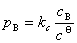
������ʽΪ�Բ�ͬ������B����ɱ�ȱ�ʾ��ϡ��Һ�����ʵĻ�ѧ�ơ�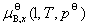 ��
��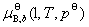 ��
��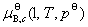 Ϊ
Ϊ ��
��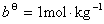 ��
��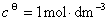 �����¶�ΪT��ѹ��Ϊʱ�����غ������ɵĻ�ѧ�ơ��ü���״̬�Ļ�ѧ��Ҳ��Ϊ��̬��ѧ�ơ�
�����¶�ΪT��ѹ��Ϊʱ�����غ������ɵĻ�ѧ�ơ��ü���״̬�Ļ�ѧ��Ҳ��Ϊ��̬��ѧ�ơ�
8��ϡ��Һ��������
ϡ��Һ������ѹ���͡����̵��½����е����ߺ���ѹ�����ʵ���ֵ����Һ���������ʵķ��ӵ���Ŀ�����ȣ�������ӵı����أ��ʳ�Ϊϡ��Һ�������ԡ�
(1) ����ѹ����
���ڶ������Һ���ܼ�������ѹ�����ɿɱ�ʾΪ

(2) ���̵��½�
��ϡ��Һ����ʱ��ֻ�������ܼ��������ʲ�����ʱ��ϡ��Һ�����̵�ȴ��ܼ������̵�ͣ��併��ֵ���������ʵ�����Ħ��Ũ�ȣ���
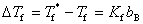
ʽ�У�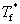 Ϊ���ܼ������̵㣻
Ϊ���ܼ������̵㣻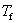 Ϊϡ��Һ�����̵㣻
Ϊϡ��Һ�����̵㣻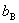 Ϊ���ʵ�����Ħ��Ũ�ȣ�
Ϊ���ʵ�����Ħ��Ũ�ȣ�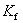 Ϊ���̵㽵�ͳ������������ܼ��������йأ���
Ϊ���̵㽵�ͳ������������ܼ��������йأ���
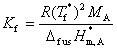
(3) �����
��ϡ��Һ�����������Dz��ӷ������ʣ���ϡ��Һ�ķе�ȴ��ܼ��ķе�������������ֵ������B������Ħ��Ũ�ȳ����ȣ���
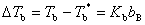
ʽ�У�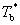 Ϊ���ܼ��ķе㣻
Ϊ���ܼ��ķе㣻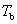 Ϊϡ��Һ�ķе㣻
Ϊϡ��Һ�ķе㣻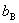 Ϊ���ʵ�����Ħ��Ũ�ȣ�
Ϊ���ʵ�����Ħ��Ũ�ȣ�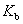 Ϊ�е����߳������������ܼ��������йأ���
Ϊ�е����߳������������ܼ��������йأ���
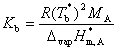
(4) ��ѹ
����ʵ��õ���ϡ��Һ����ѹ����Һ����������B����Ũ�ȳ����ȣ���
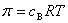
ʽ�У���Ϊϡ��Һ����ѹ��cBΪ����B����Ũ�ȡ�
9. ���䶨��
(1) ���䶨��
�ڵ��µ�ѹ�����£����һ��������ͬʱ�ܽ�����������Ļ������ܵ�Һ����ﵽ����ƽ��ʱ���������������е�Ũ��֮��Ϊһ����������
��������������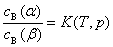
ʽ��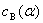 ��
��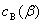 �ֱ��ʾ����B��
�ֱ��ʾ����B�� Һ���
Һ���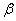 Һ���е�Ũ�ȣ�K��T��p����Ϊ����ϵ����
Һ���е�Ũ�ȣ�K��T��p����Ϊ����ϵ����
(2) ���䶨�ɵ�Ӧ��
����n����ȡ����ȡ���������ʵ�����Ϊ

��4����ѧƽ��
1����ѧ��Ӧƽ������
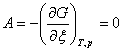 ��
�� 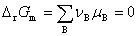
2����ѧ��Ӧ���·���ʽ�ͻ�ѧ��Ӧ������ж�
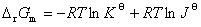
�ڵ��µ�ѹ�ͷ������Ϊ�������£��Է��ϵͳ�н��еĻ�ѧ��Ӧ��
�� ��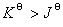 ����
���� 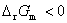 ����Ӧ�������Է����У�
����Ӧ�������Է����У�
�� ��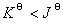 ����
���� 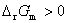 ����Ӧ�������Է����У�
����Ӧ�������Է����У�
�� ��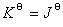 ����
���� 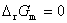 ����ʾϵͳ�Ѵ���ƽ��״̬��
����ʾϵͳ�Ѵ���ƽ��״̬��
3. ƽ�ⳣ���ĸ��ֱ�ʾ�����ͼ���
(1) ��ƽ�ⳣ���;���ƽ�ⳣ�������������巴ӦϵͳΪ����
û�����١�
(2) ����ƽ�ⳣ����ʾ��
�� ��ƽ���ѹ��ʾ��ƽ�ⳣ��
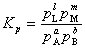 ��
�� 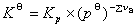
�� �����ʵ���������ʾ��ƽ�ⳣ��
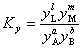 ��
�� 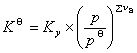
�� ����Ũ�ȱ�ʾ��ƽ�ⳣ��
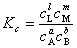 ��
�� 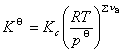
�� �����ʵ�����ʾ��ƽ�ⳣ��
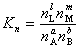 ��
�� 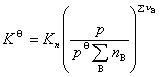
(3) ƽ�ⳣ���ļ���
�� ��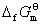 ����ƽ�ⳣ��
����ƽ�ⳣ�� 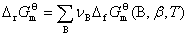
�� ����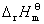 ��
��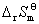 ����ƽ�ⳣ��
����ƽ�ⳣ�� 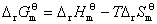
�� ���ü����йػ�ѧ��Ӧ�� ֵ����ƽ�ⳣ��
ֵ����ƽ�ⳣ��
�����������ַ��������������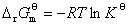 ��ϵ��
��ϵ�� ��
��
(4) ƽ�ⳣ���IJⶨ
ͨ���ⶨƽ���ϵͳ�и���ֵ�ƽ��Ũ�Ȼ����ֵķ�ѹ����ƽ�ⳣ����ϵʽ�����ƽ�ⳣ�������õIJⶨƽ��Ũ�ȵķ������������ͻ�ѧ����
4. ƽ��ת���ʺ�ƽ����ʵļ���


5. �������ضԻ�ѧƽ���Ӱ��
(1) �¶ȶԻ�ѧƽ���Ӱ������ѧ��Ӧ��ѹ����ʽ
�� ��������ʽ 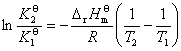
�� ����������ʽ 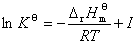
�� 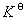 ���¶�T�ĺ�����ϵ
���¶�T�ĺ�����ϵ
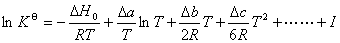
(2) ѹ���Ի�ѧƽ���Ӱ��
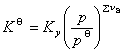
��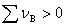 ʱ���� p������
ʱ���� p������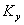 ��С������ѹ����������ķ�Ӧ������
��С������ѹ����������ķ�Ӧ������
��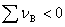 ʱ����p��������������ѹ���������С�ķ�Ӧ������
ʱ����p��������������ѹ���������С�ķ�Ӧ������
��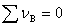 ʱ���� p���������䣬����ƽ����Ӱ�졣
ʱ���� p���������䣬����ƽ����Ӱ�졣
(3) ��������Ի�ѧƽ���Ӱ��
��ѹһ��ʱ����������Ĵ���ʵ��������ϡ�����ã����ͼ��ٷ�Ӧϵͳ��ѹ��Ч����ͬ��
(4) ��Ӧ����ȶԻ�ѧƽ���Ӱ��
��ѧ��Ӧ�иı䷴Ӧ�����ȣ���һ�ּ�����ԭ���ʵ�����������Ӧ��ƽ��ʱ�����������һ��ԭ�ϵ�ת���ʡ�
��5����ƽ��
1���������������
(1) �ࡢ������������������ɶȵĶ��塣
(2) ���ɵij�����ѧ����ʽ ?��K������2��ʽ��2��ָ�¶Ⱥ�ѹ������������
2�������ϵͳ
(1) ��ͼ ָ����ʵ�����������Ƴ���ƽ��ϵͳ�Ĵ���״̬ͼ��
(2) ��ͼ���������۵ķ����Ͳ��� �� ȷ����ͼ�����ͣ��ڷ�����ͼ�������ֵĵ㡢�ߡ��棻����ͼ�пɱ�������ϵͳ״̬�仯֮��Ĺ�ϵ������ͼ��Ӧ�á�
(3) ˮ����ͼ ��Ϊ������������lʱ��?��2��ʵ��Ϊ����ƽ���ߣ�����2ʱ��?��1��O��Ϊ����㣬����3ʱ��?��0��ע�������ͱ��������
3�������ϵͳ
(1) ������ϵ�㣺�ֱ��ʾϵͳ��һ����������ͼ������ɻ�����ɵĵ㡣
(2) �ܸ˹��� ����˶����ϵͳ�����ʱ�������������ļ������⣬�������ڶ���������������ϵͳ�������Ħ��������ʾʱ������������֮��ΪĦ����֮�ȣ���ϵͳ�����������������ʾʱ������������֮��Ϊ����֮�ȡ�
(3) �������-Һƽ��ϵͳ
�� ������Һϵͳ p-��ͼ��p-x-y��ͼ��T-x-y��ͼ
�� ��������Һ ���ϵͳT-x-y��ͼ
�� �������뾫��Ļ���ԭ��
�� ˮ��������ԭ��
(4) �����Һ-Һƽ��ϵͳ
�پ�������ٽ��ܽ��¶ȵ�ϵͳ
�ھ�������ٽ��ܽ��¶ȵ�ϵͳ
�� ͬʱ�����ϡ����ٽ��ܽ��¶ȵ�ϵͳ
�ܲ������ٽ��ܽ��¶ȵ�ϵͳ��
(5)�����Һ-��ƽ��ϵͳ
�� ����ϵͳ
�� ���ɻ�����Ķ����ϵͳ
4. �����ϵͳ
(1) �����ϵͳ�ȱ������α�ʾ����
(2) �����ϵͳ��ͼ
�� ����־���һ�Բ��ֻ��ܵ�Һ-Һϵͳ �� �����ˮ��ϵͳ��
��6�� ������ѧ��Ӧ����ѧ
1. ��ѧ��Ӧ���ʵĶ���
(1) �Է�Ӧ���ȱ�ʾ��Ӧ����
���л�ѧ��Ӧ�����������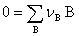 ���û�ѧ��Ӧ������Ϊ��
���û�ѧ��Ӧ������Ϊ��
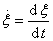
�����뷴Ӧ��������Ϊ ʱ������
ʱ������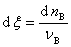 ����ʽ�ɸ�дΪ
����ʽ�ɸ�дΪ

ʽ�У��� Ϊ��Ӧ���ȣ�tΪʱ�䡣
��2�����ݷ�Ӧ�ķ�Ӧ����
���ں��ݷ�Ӧ����Ӧϵͳ�����V����ʱ��仯����B��������Ũ��Ϊ ��������
��������
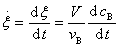
���� 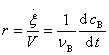
��ʽ��Ϊ���ݷ�Ӧ�ķ�Ӧ���ʳ��ö��塣
����ʽ�����ڷ�Ӧ ������
������
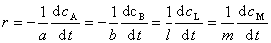
ʽ�У�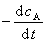 ��
��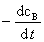 �ֱ��Ϊ��Ӧ�� A��B ���������ʣ�Ҳ������
�ֱ��Ϊ��Ӧ�� A��B ���������ʣ�Ҳ������ ��
��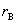 ��ʾ��
��ʾ��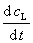 ��
��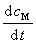 �ֱ��Ϊ���� L��M ���������ʣ�Ҳ������
�ֱ��Ϊ���� L��M ���������ʣ�Ҳ������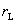 ��
��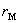 ��ʾ����ʵ��Ӧ���У�������
��ʾ����ʵ��Ӧ���У������� ��������ʾ��Ӧ�ķ�Ӧ���ʡ�
��������ʾ��Ӧ�ķ�Ӧ���ʡ�
2. ��Ӧ������Ũ�ȵĹ�ϵ
(1) ��Ӧ������Ũ�ȹ�ϵ�ľ��鷽��
���ڷ�Ӧ
ͨ��ʵ������Ӧ������Ũ�ȵĹ�ϵΪ
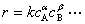
ʽ�� �������ֱ�Ϊ A��B ���ʵķ�Ӧ�ּ������� ��+��= n �Ʒ�Ӧ���ܼ�����
(2) ��Ӧ���ʵ�����ʽ�ͻ�����ʽ
1) һ����Ӧ
��ʵ��ȷ��ij��Ӧ��A�����������뷴Ӧ��Ũ��һ�η������ȣ���Ϊһ����Ӧ���������ʷ���Ϊ
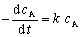
��������ʷ���Ϊ 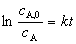
һ����Ӧ�Ķ���ѧ����Ϊ��
�� ��Ӧ���ʳ���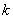 �ĵ�λΪʱ��-1���� ��Ӧ��A�İ�˥��
�ĵ�λΪʱ��-1���� ��Ӧ��A�İ�˥��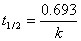 �����Ũ��
�����Ũ��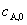 �أ���
�أ��� 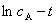 ͼΪһ��ֱ�ߡ�
ͼΪһ��ֱ�ߡ�
2) ������Ӧ
�� ֻ��һ�ַ�Ӧ������
��ʵ��ȷ��ij��Ӧ��A�����������뷴Ӧ��Ũ�ȶ��η������ȣ���Ϊ������Ӧ���������ʷ���Ϊ
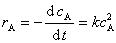
��������ʷ���Ϊ 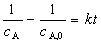
�� �����ַ�Ӧ������
�練Ӧ
��ʵ��ȷ��ij��Ӧ��A�����������뷴Ӧ��A��B����Ũ��һ�η������ȣ����ܷ�ӦΪ�������������ʷ���Ϊ
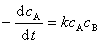
��������ʷ���Ϊ 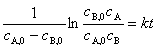
ֻ��һ�ַ�Ӧ��Ķ�����Ӧ�Ķ���ѧ����Ϊ��
(��) ������Ӧ���ʳ��� k�ĵ�λΪŨ��-1·ʱ��-1��(��) ��Ӧ��A��˥��Ϊ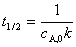 �뷴Ӧ���ʼŨ�ȳɷ��ȣ�(��)
�뷴Ӧ���ʼŨ�ȳɷ��ȣ�(��) 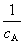 ��t��ͼΪһ��ֱ�ߡ�
��t��ͼΪһ��ֱ�ߡ�
3) n����Ӧ
��Ӧ 
����Aʱn����Ӧ�����������ʷ���Ϊ
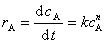
��������ʷ���Ϊ
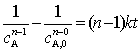 ��
��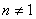 ��
��
n����Ӧ�İ�˥��Ϊ
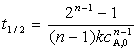 ����
����
3. ��Ӧ����ȷ���ķ���
(1) ���ַ�
��ʵ�����ݴ���������ķ�Ӧ����ʽ����� k���� k Ϊ������Ϊ�˼��������������Թ�ϵͼ������ֱ����Ϊ�ü������˷�����������������
(2) �ַ�
��  ��ʽ�Ķ���ѧ���̣�
��ʽ�Ķ���ѧ���̣� ����lgrA��lgk��ֱ�߹�ϵ���ؾ�Ϊn��������
����lgrA��lgk��ֱ�߹�ϵ���ؾ�Ϊn�������� ���м��㡣��ʱ
���м��㡣��ʱ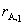 ��
��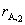 ���ʵ��õ� cA�� tͼ��������˷������������������������ó�ʼŨ�ȷ����ɱ������ĸ��š�
���ʵ��õ� cA�� tͼ��������˷������������������������ó�ʼŨ�ȷ����ɱ������ĸ��š�
(3) ��˥�ڷ�
��һ����Ӧ�⣬�����������ͬ��ʼŨ���� �������飬�ֱ�ð�˥��Ϊ
�������飬�ֱ�ð�˥��Ϊ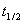 ��
��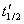 ������
������
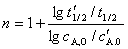
(4) ���뷨
���������ֻ��������Ϸ�Ӧ��ķ�Ӧ����  �����㶨 cB��
�����㶨 cB��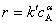 ����ͨ�����������������ͬ���㶨 A Ũ��ʱ
����ͨ�����������������ͬ���㶨 A Ũ��ʱ 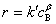 �����������
�����������
4����Ԫ��Ӧ���������ö���
(1) ��Ԫ��Ӧ
��ָ�ɷ�Ӧ���������ӡ�ԭ�ӡ��������ɻ��ȣ�һ��ֱ��ת��Ϊ����ķ�Ӧ������Ӧ�����������������ϵĻ�Ԫ��Ӧ����ɣ���÷�Ӧ��Ϊ�ǻ�Ԫ��Ӧ����Ӧ����������ij��Ԫ��Ӧ�ķ������Ʒ�Ӧ����������Ӧ������ֻ��������������Ӧ��Ϊ�����ӡ�˫���Ӻ������ӷ�Ӧ��
(2) �������ö���
�Ի�Ԫ��Ӧ����Ӧ���������Ӧ��Ũ�ȵ��ݳ˻������ȣ����и�Ũ�ȵķ���Ϊ��Ӧ�����и���Ӧ��Ļ�ѧ������������

�����ʷ���Ϊ
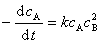
ע�⣺�� �Էǻ�Ԫ��Ӧ�������������ö��ɡ��ڷ�Ӧ�����뷴Ӧ��Ũ�ȳ˻������ȣ����������Ũ�ȡ�
5. �¶ȶԷ�Ӧ���ʵ�Ӱ��
(1) ���ػ���Van��t Hoff������

(2) ��������˹��Arrhenius������
����ʽ  ָ����ʽ
ָ����ʽ 
��������ʽ  �� ln k�� lg k��
�� ln k�� lg k��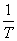 ��ͼΪһֱ�ߣ���ֱ��б�ʺͽؾ࣬�ɷֱ������� Ea��ָǰ����A��
��ͼΪһֱ�ߣ���ֱ��б�ʺͽؾ࣬�ɷֱ������� Ea��ָǰ����A��
������ʽ 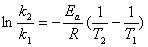
6. ��Ӧ�������۵Ļ�����ʽ
(1) ����ײ����
�� ����ײ���۵Ļ�������
(��) ��������Ǹ�����״���ӣ�������A�ͷ���B������Ӧʱ���Ⱦ������DZ��뾭����ײ��(��) ֻ����Щ����һ����ӵ�ƽ�������Ļ���Ӽ����ײ������ײ����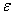 ���ڻ�������ٽ���c�����ܣ�����ײ������������һ���ռ����õļ�������ʱ�����ܷ�����ѧ��Ӧ������)�ڷ�Ӧ�����У�������ӵ��������DZ�����ƽ��ķֲ���
���ڻ�������ٽ���c�����ܣ�����ײ������������һ���ռ����õļ�������ʱ�����ܷ�����ѧ��Ӧ������)�ڷ�Ӧ�����У�������ӵ��������DZ�����ƽ��ķֲ���
�� ����ײ���۵Ļ�����ʽ
(��)ͬ���ӷ�Ӧ
��ײƵ�� 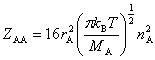
���ʳ��� 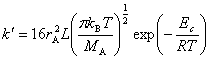
(��) �������
��ײƵ�� 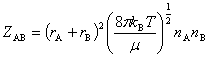
���ʳ��� 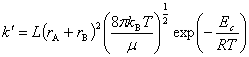
(2) ����״̬����
�� ��Ӧģʽ
A + BC  ��A��B��C��
��A��B��C�� AB + C
AB + C
ʽ��A��B��C��ʾ����ԭ�ӣ�BC��AB��ʾ���ӣ���A��B��C�ݱ�ʾ�����
�� ����״̬���۵Ļ�����ʽ
(��) ͳ������ѧ����ʽ
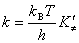
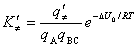
(��) ����ѧ����ʽ
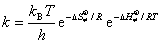
ʽ��  ��
�� �ٷֱ�Ϊ��غͻ�ʡ�
�ٷֱ�Ϊ��غͻ�ʡ�
��7�����ӷ�Ӧ�����ⷴӦ����ѧ
1. �����ӷ�Ӧ
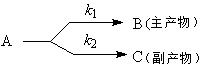
(1) ƽ�з�Ӧ
��һ�ֻ��ַ�Ӧ��μӵ���ͬ������е�ͬʱ���ڵķ�Ӧ��ƽ�з�Ӧ����
���ʷ��̵Ļ���ʽ 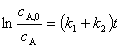
�� 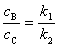
����Ӧ��������ͬ������Ԫ��Ӧ��ƽ�з�Ӧ������������ӦŨ��ֵ�ȵ������ʳ���֮�ȡ���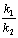 ���������¶�һ��ʱ��Ϊ��ֵ���������ij����IJ�������Ҫ��ı��ı�ֵ��
���������¶�һ��ʱ��Ϊ��ֵ���������ij����IJ�������Ҫ��ı��ı�ֵ��
(2) ���ŷ�Ӧ
�����淽��ͬʱ���еķ�Ӧ�϶��ŷ�Ӧ���ֳƿ��淴Ӧ����

���ʷ��̵Ļ���ʽ 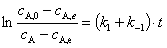
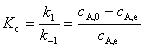
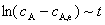 ��ֱ�߹�ϵ����ֱ��б�ʿ���ã�k1��k-1��������ƽ�ⳣ�� Kc���
��ֱ�߹�ϵ����ֱ��б�ʿ���ã�k1��k-1��������ƽ�ⳣ�� Kc��� 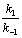 ������ʽ����������ͬʱ���k1��k-1��
������ʽ����������ͬʱ���k1��k-1��
(3) һ��������Ӧ
��һ����Ӧ�IJ��ֻ�ȫ������������һ����Ӧ�IJ��ֻ�ȫ����Ӧ��ʱ�ķ�Ӧ��Ϊ������Ӧ����
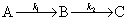
��A��B��C��Ũ�ȱ�ʾ�����ʷ���Ϊ
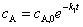
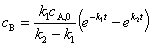
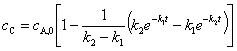
���м����B����������Ҫ�ģ���cB�����ֵ����ѷ�Ӧʱ����
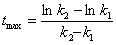
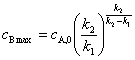
2�����ӷ�Ӧ���ʵĽ��ƴ�����
(1) ѡȡ���Ʋ��跨
����������Ӧ��������ijһ����Ӧ�������������ܵķ�Ӧ�������õ�����һ����Ϊ��Ӧ���ʵĿ��Ʋ��衣����������Ӧ
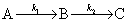
����һ����Ӧ�����ʺ������ڶ�����Ӧ���ʺܿ죬��Ӧ��������
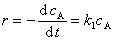
(2) ��̬���Ʒ�
��Ӧ A + B  C + D
C + D
���з�Ӧ������MΪ�м��
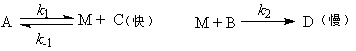
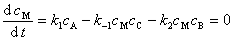
(3) ƽ��̬���Ʒ�
�ٶ���Ӧ�����а�����һ�����淴Ӧ���ڿ��淴Ӧ֮�������һ���ٶȽ��������ʾ������裬��
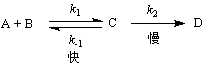
�������һ��Ϊ�����裬���ǰ��Ŀ��淴Ӧ����ʱά��ƽ�⣬��
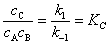 ��
��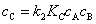
3������Ӧ
(1) ����Ӧ�Ĺ�ͬ����
����Ӧһ��Ӧ������������ɣ���������������������������ֹ��
��2������Ӧ����
����Ӧ��Ϊ��ֱ����Ӧ��֧����Ӧ��ǰ��ÿ����һ���ʵ�֮����һ���µĻ����ʵ㣻����ÿ����һ�������ʵ�ͬʱ������������������ϵĻ����ʵ㡣
4������Ӧ
(1) ������μӿ췴Ӧ����
ԭ���Ͻ����ӷ�Ӧ��仯���������������;��ʵ�֣�����ѧ��Ӧ���Ż�ܽϵ͵�;�����С�����֮�����ܼӿ췴Ӧ���ʣ������ڸı��˷�Ӧ;���������˷�Ӧ�Ļ�ܣ�����������Ƶ�����ӡ�
(2) ����������
�� ��Ӱ�컯ѧƽ�⡣
�� �����Է�Ӧ�ļ�����ѡ���ԡ�
�� �����ڷ�Ӧǰ��ѧ���ʲ��䣬�������������ʵı仯��
�� �����У���Ӧϵͳ�У����ü����������ʶ�ʹ��������ǿ�ͣ�ǰ���������������ã�����������ʹ�������ж�����
(3) �������Ӧ
ָ��Ӧ���������ͬһ���н��з�Ӧ���������Ӧ���ص��Ƿ�Ӧ��ʹ����ܹ���־��ȽӴ������Լ�ѡ���Խϸߣ���Ӧ�����ºͣ��������ķ���ͻ��ս�Ϊ���ѡ������������
�� �����������Ϊ���壬��NO��g����H2O��g���������������Ӧ��������Ӧ������
�� ��������Ҫ���������ӵ�ת�ơ�
�� ��ϴ���һ���þ��н�ǿ��������Ĺ��ɽ�����������ͨ�������������ʹ��Ӧ�������ڷ�Ӧ��
(4) �������Ӧ�IJ���
�� ��Ӧ��ӱ�����ɢ���������棻
�� ��Ӧ���ڴ�������������
�� ��Ӧ���ڴ������淢����Ӧ���ɲ��
�� ����Ӵ��������Ѹ���
�� �Ѹ��������ɢ���������С�
���岽���������еģ�������Ӧ������������������һ��������
(5) ø����Ӧ���ص�
�� ��Ч�ʸߣ��� ��Ӧ�����ºͣ� �� �߶�ѡ���ԡ�
��8���������Һ����
1�������ڵ�ⶨ��
������ͨ���������Һʱ��ͨ���缫�ĵ����뷢���缫��Ӧ�����ʵ��������ȣ���
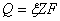
2�����ӵ�Ǩ����
ͨ��ʱ���������Һ��ij������Ǩ�Ƶĵ������ܵ���֮�ȳ�Ϊ���ӵ�Ǩ��������t��ʾ����
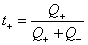
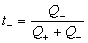
3���絼���絼�ʺ�Ħ���絼��
(1) �絼���絼��
���ȵ����ھ��ȵ糡�еĵ絼G�뵼������A�����ȣ��볤��l�ɷ��ȣ���
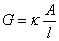
ʽ�У� Ϊ�絼�ʣ��䵥λΪ
Ϊ�絼�ʣ��䵥λΪ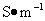 ��
��
(2) Ħ���絼��
Ħ���絼����ָ���Ϊ1m����ƽ�е缫֮����ú���1mol����ʵ���Һ������Һ�ĵ絼�ʳ�ΪĦ���絼�ʣ�����m��ʾ����

4�����Ӷ����˶�����
������ϡ�͵ĵ������Һ�У����е����ȫ�����룬�������ӻ���Ӱ�죬�˴˶����˶���ÿ�����ӶԵ������Һ�ĵ��������Ĺ��㶨���䣬�������Һ�ļ���Ħ���絼���Ǹ����ӵĵ絼��֮�͡�
 5�����ӵ�ƽ�������ƽ�����ϵ��
5�����ӵ�ƽ�������ƽ�����ϵ��
5. �������Һ�Ļ�ȺͻҶ�ϵ��
(1) ����ʺ����ӵĻ�ѧ�Ƽ����
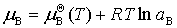
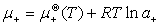

����
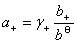
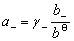
ʽ��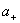 ��
�� ��
��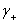 ��
��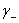 ��
��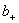 ��
��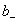 �ֱ�Ϊ���������ӵĻ�ȡ����ϵ��������Ħ��Ũ�ȡ�
�ֱ�Ϊ���������ӵĻ�ȡ����ϵ��������Ħ��Ũ�ȡ�
(2) ƽ�������ƽ�����ϵ��
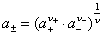
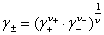
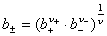
ʽ��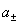 ��
��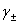 ��
�� �ֱ�Ϊƽ����ȡ�ƽ�����ϵ����ƽ������Ħ��Ũ�ȡ�
�ֱ�Ϊƽ����ȡ�ƽ�����ϵ����ƽ������Ħ��Ũ�ȡ�
6���°�-�ݿ˶�������
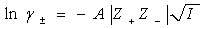
���� 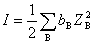
��9����صĵ綯���뼫������
1. ԭ�������ص��
��ؿ���Ϊ��ԭ��غ͵��ص��ܳơ�ԭ����ǰѻ�ѧ��ת��Ϊ���ܵ�װ�ã��������ǰѵ���ת��Ϊ��ѧ�ܵ�װ�á�
������������缫֯�ɵģ��������缫�Ϸֱ����������Ӧ�ͻ�ԭ��Ӧ����Ϊ�缫��Ӧ�������缫��Ӧ���ܽ����Ϊ��ط�Ӧ���绯ѧ�й涨������������Ӧ�ĵ缫��Ϊ������������ԭ��Ӧ�ĵ缫��Ϊ�������֣�������Ϊ��Դ�еĵ缫���Ƶĸߵͣ��涨���Ƹߵĵ缫��Ϊ���������Ƶ͵ĵ缫��Ϊ������
2���缫������
(1) ��һ��缫�����������缫������缫��
�� �����缫: �ɽ������뺬�иý������ӵ���Һ�������ɡ�����Mn+|M
�� ����缫: �ɶ��Խ���(ͨ����Pt��Au)��������ͺ��и����������ӵ���Һ�й��ɡ�����缫�� Pt|H2{p(H2)}|H+{a(H+)}
(2) �ڶ���缫: ��������-�����ε缫�ͽ���-����������缫
�� ����-�����ε缫���磬��-�Ȼ����缫Ag-AgCl��Cl-{a(Cl-)}���ʹ��缫�缫Hg(l)��Hg2Cl2(s)��Cl-{a(Cl-)����
�� ����-����������磬��-�������缫�����Ի���Hg��HgO��H+(a)�����Ի���Hg��HgO��OH+(a)
(3) ������缫: ��������ԭ�缫��
�����Ե缫���뺬��ͬһԪ�ص����ֲ�ͬ��̬���ӵ���Һ�м����ɣ���Pt��Fe2+��Fe3
(4) ������缫������ѡ���Ե缫��
3��ԭ��صĵ綯�Ƽ���˹�ط���
(1) ԭ��ص綯�ƵĶ���
��û�е���ͨ���������£�ԭ��ص������Ľ�������Ϊͬ�ֽ���ʱ��������˵��Ʋ��Ϊԭ��صĵ綯�ƣ���E��ʾ����
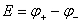
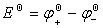
(2) ������
�����ر���߱���������������
�� ����ڽ��еĻ�ѧ��Ӧ�����ǿ���ģ�
�� ����ڳ䡢�ŵ�Ĺ���������ת�����棻
�� ����н��е���������Ҳ������档
(3) ��ط�Ӧ����˹�ط���
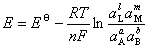
4���綯�ƲⶨӦ��
(1) �����ط�Ӧ�й�����ѧ�����ı仯ֵ���� ��
��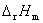 ��
��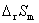 ��
��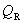 ��
��
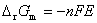
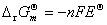
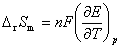
ʽ�� Ϊ��ص綯�Ƶ��¶�ϵ����
Ϊ��ص綯�Ƶ��¶�ϵ����
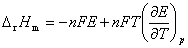

(2) �ɼ����ط�Ӧ�ı�ƽ�ⳣ�������볣���������ε��ܶȻ���ˮ�����ӻ���
�� ��ƽ�ⳣ������綯�ƵĹ�ϵ 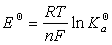
�� �ܶȻ������ļ��� 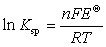
(3) �ɵIJⶨ�ɼ�����������ƽ�����ϵ����pHֵ
�� ���ϵ��������
�� pHֵ�ļ���
��10��Һ��ı�������
1���ȱ���
��λ��������������еı��������Ϊ�ȱ��棬�Է���AV��ʾ��
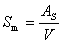
���ʷָ����ϸ�ȱ���������ֳ��ı���ЧӦ��ǿ��
2�����湦
�����ʹ���������dA�����ϵͳ���Ĺ����������湦��
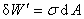
��������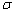 ��ָ��T��p����ɺ㶨�������£����ӵ�λ�����ʱ�������ϵͳ���Ŀ�����������
��ָ��T��p����ɺ㶨�������£����ӵ�λ�����ʱ�������ϵͳ���Ŀ�����������
3���ȱ��漪��˹������
������T��p����ɺ㶨�������£����ӵ�λ�����ʱ�����ı��湦����ϵͳ����˹�����ܵı仯��
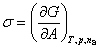
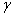 ��Ϊ�ȱ��漪��˹�����ܣ��䵥λΪJ?m-2��Ҳ������Ϊ��λ������ϵķ��ӱ��ڲ����Ӿ��и��ߵ�������������������Ϊ���漪��˹�����ܡ�
��Ϊ�ȱ��漪��˹�����ܣ��䵥λΪJ?m-2��Ҳ������Ϊ��λ������ϵķ��ӱ��ڲ����Ӿ��и��ߵ�������������������Ϊ���漪��˹�����ܡ�
4����������
Һ��������һ��ʹҺ���Զ�������������Ϊ�������������ڱ��������Ĵ��ڣ�����Һ��һ������Ρ�
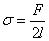
Һ������ϴ�ֱ�����ڵ�λ�����߶��ϵı������������Ϊ�������������漪��˹�����ܺͱ���������ͬһ��������ֲ�ͬ���﷽ʽ����Ȼ�������岻ͬ�������Ǿ��еȼ۵����ٺ���ͬ����ֵ��
��Һ��Ϊƽ�棬���������ķ�����Һ��ƽ�У���Ϊ���棬���������ķ�����Һ������߷���һ�¡�
5������Һ���µĸ���ѹ��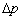
ƽҺ�棬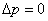 ��
��
Һ�棬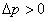 ��
��
��Һ�棬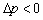 ��
��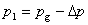
���ӵ�ѹ���ķ���ָ����������ġ�
6��������˹��ʽ
������˹��ʽ�����˸���ѹ�������ʰ뾶�Ĺ�ϵ��
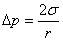 �����棩
�����棩
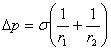 ���������棩
���������棩
7��ëϸ����
��Һ������ʪëϸ�ܣ�Һ�������ʰ��棻��Һ�岻����ʪëϸ�ܣ���Һ���½����档�������½��ĸ߶ȿ�ͨ����ʽ���㡣
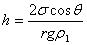
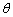 ΪҺ����ܱ�֮��ĽӴ��ǡ�
ΪҺ����ܱ�֮��ĽӴ��ǡ�
8�������ķ���
�����ķ���������Һ�������ѹ�����ʰ뾶֮��Ĺ�ϵ��
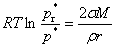
����Һ�Σ��뾶��С������ѹ�����������ݣ��뾶��С��Һ�������ڵ�����ѹ���͡�
9������˹��������ʽ
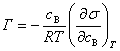
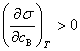 ��
��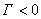 �������ڱ�����Ũ��С������Ũ�ȣ��Ǹ�������
�������ڱ�����Ũ��С������Ũ�ȣ��Ǹ�������
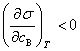 ��
��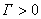 �������ڱ�����Ũ�ȴ�������Ũ�ȣ�����������
�������ڱ�����Ũ�ȴ�������Ũ�ȣ�����������
10������������
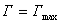
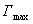 ��Ϊ������������������������ɱ�������������õ�����Ĥ�еķ��ӽ����A��
��Ϊ������������������������ɱ�������������õ�����Ĥ�еķ��ӽ����A��
11��������Լ�HLBֵ�ļ���
��ά˹��ֵ����
����ұ�ֵ����
��11�������������
1������������ʪ����
(1) ��ʪ����������ϵ����壨��Һ�壩��Һ�壨����һ��Һ�壩ȡ��������
(2) ��ʪ�����ͣ�մ����ʪ��������ʪ����չ��ʪ��
����������ʪ�����ܷ��Է����У��ɺ��º�ѹ�µļ���˹�������о���������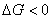 ������ʪ��
������ʪ��
մʪ�� 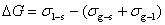
��ʪ��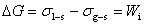
��չ��
(3) ��չϵ��
��S>0��Һ�����������չ�ڹ�����档
(4) �Ӵ��Ǻ���ʪ����
�� �Ӵ��ǣ�������Һ�������ཻ�紦����Һ���澭��Һ���ڲ�����Һ����֮��ļнǽ����Ӵ��ǣ�������ʾ��
�� ��ʪ���� 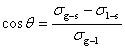
ϰ���ϣ�����90°Ϊ��ʪ������90°Ϊ����ʪ���Ӵ��ǵ���0°��Ϊ��չ��
2����������ϵ�����
(1) ���������뻯ѧ����
ͨ������������ڹ����������Ծۼ����ܶ�����������Ϊ�����ڹ�������ϵ��������������������Ϊ�����ʣ��������������Ĺ����Ϊ��������
���������������ı��ʲ�ͬ���ɷ�Ϊ���������ͻ�ѧ������ǰ��Ϊ���»���������Ϊ��ѧ������
(2) ���ֵ����������·���ʽ
���ֵ���ϣͨ������ʵ�����ݹ����ܽ����������������һ�����鷽�̣��磺

3���ʸ��Ѷ���������������
(1) ����Ҫ��
�� ������������ķ����ǵ����Ӳ�ģ��ڹ��������������Ǿ��ȵģ��۱�������������Ӽ�����������������ƽ���Ƕ�̬ƽ�⡣
(2) ���·���ʽ�� 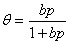
(3) ������������һ�����ӱ�����ʱ������������ӣ�����ռһ���������ģ���Ϊ��������������·���ʽΪ
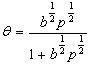
(4) ����������������к���A��B�������壬�Ҿ��ܱ���������������A�����ڱ����Ϸ�����Ӧ�����ɵIJ���BҲ�ܱ���������Щ��������Ϊ�ǻ������������·���ʽΪ
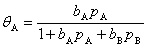
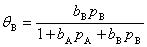
4������Ӳ���������
(1) ����Ҫ��
�� ����ɷ�������Ӳ��������� ���˵�һ��������Ϊͨ��˵���������⣬��������������Ⱦ�Ϊ����(��������)��Һ���ȣ��� ����(��������)���ӵ�����������ֻ�����ڱ�¶������ı����ϣ��� �����ﵽ��̬ƽ��ʱ�������������������ֲ���
(2) ���·���ʽ
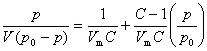
ʽ��p0Ϊ��������ʱ�ı�������ѹ��
5����������Һ������
(1) �������ļ���
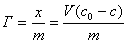
��2�����·���ʽ
�� ���ֵ����������·���ʽ 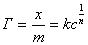
�� ����Ӳ��������·���ʽ 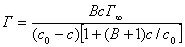
(3) Ӱ������
Ӱ�����������ذ��������������ܼ������������Բ�ͬ����������Ӱ�죻�������ܼ��е��ܽ�ȶ���������Ӱ�죻�¶ȶ���������Ӱ�죻�������ı���״̬����������Ӱ��
��12�������ɢϵͳ
1����ɢϵͳ
(1) ��ɢϵͳ�Ķ���
һ�ֻ������ʷ�ɢ����һ�������������ɵ�ϵͳ�з�ɢϵͳ������ɢ�����ʽз�ɢ�ࣻ���ɢ���õ����ʽз�ɢ���ʡ�
(2) �����ɢϵͳ�ʹַ�ɢϵͳ
����ɢ�����Ӵ�С��1~100nm֮���Ϊ�����ɢϵͳ�������ܽ�����Һ���壩�����ţ��������ʣ����������Һ����Һ���壩�������Ӵ�С����100nm��Ϊ�ַ�ɢϵͳ��������״Һ����ĭ�������塣
2��������Ʊ��봿��
(1) 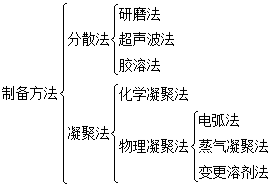 ��2��
��2��
3. �ܽ�����Ҫ����
�ܽ�����Ҫ�����ǣ��߶ȷ�ɢ�ģ���ɢ����1nm~100nm��������ģ������ȵģ�������ѧ���ȶ�ϵͳ��
4. ����Ķ�������
(1) �����˶�
�ܽ��еķ�ɢ�����������ܵ���������˷��������˶��ķ�ɢ���ʵ�ײ��������������˶��в����˶��������˶������������ɢ�������ܽ�����Ҫ��������֮һ��
�ɲ����˶��ɵõ������ڹ۲�ʱ���ڵIJ����˶�λ�ƹ�ʽ������
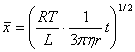
(2) ��ɢ����
����Ũ���ݶȵĴ����������ӴӸ�Ũ��(�����������)�������Ũ��(�����������)������Ǩ�Ƶ��������ɢ��
��ɢ��ӷƿ���ɢ���ɣ���
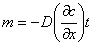
(3) ���������ƽ��
�ܽ��еķ�ɢ�������������������������������÷����³��Ĺ��̳�Ϊ������
�� ���������з�ɢ���ڷ�ɢ�����еij�����������ʽ��ʾ��
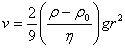
��ɢ�����ӱ���������ʹ���ӳ����������ʵ�ճ�ȼ������˶��������ɢ������ֹ�����³������������൱ʱ�ﵽƽ�⣬��֮Ϊ����ƽ�⡣
��Ӧ�ó���ƽ��ԭ��������ϵͳŨ��(���������)�ĸ߶ȷֲ���
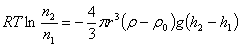
�� �ڳ����ij��з�ɢ���ڷ�ɢ�����еij�����ʽ����ʽ��ʾ
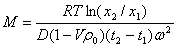
����ƽ��ʱ�Ĺ�ʽ��
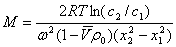
5���ܽ���ѧ����
(1) ������ЧӦ
�����ܽ��Ĺ�ѧ�������ԡ�����һ�����������ܽ���ɢ�����ӳߴ��������յ��ܽ�ϵͳ���ɷ���ɢ������һ����������
(2) ��ɢ�乫ʽ
ɢ����ǿ�ȿ��ö���(Rayleigh)��ʽ��ʾ��
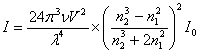
6���ܽ��ĵ�ѧ����
(1) ��������˫���
��������������뼫�Խ��ʽӴ����ڽ����ϻ���磬�Ӷ��γ�˫��㡣 ��������������ɣ���˫������Һһ����������ɣ���һ��Ϊ�����ڹ�������ˮ�������Ӳ�(����������������෴)����Ϊ˹�ض��㣬�ɳ�Ϊ���ܲ㡣�ڶ���Ϊ��ɢ�㡣�ɹ����������Һ����ĵ��Ʋ� ������ѧ���ƣ���˹�ض�������Һ�����ĵ��Ʋ�
������ѧ���ƣ���˹�ض�������Һ�����ĵ��Ʋ�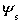 ��˹��˼���ƣ����ɻ���������Һ�������ĵ��Ʋ��
��˹��˼���ƣ����ɻ���������Һ�������ĵ��Ʋ��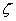 ���ƣ���е綯���ơ�
���ƣ���е綯���ơ�
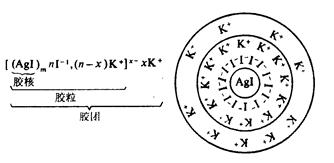
(2) ���ŵĽṹ
��AgNO3��Һ�μ���KI��Һ���γɵ�AgI�ܽ�Ϊ�����佺�Žṹ������ͼ��ʾ��
(3) �綯����
���ڽ����Ǵ���ģ������ڵ糡�����£��������ѹ������������������������ʱ�����ĵ綯�����ֳ��ܽ��ĵ�ѧ���ʡ���Щ���ʰ�������Ӿ�������� �������ƣ��������ơ�
7���ܽ����ȶ���
(1) �ܽ��Ķ����ȶ���
�����ܽ��з�ɢ�����ӵIJ����˶��ڷ�ɢ�����в�ͣ��������Ǩ�ƶ�����һ��ʱ���ڱ����ܽ��ȶ����ڵ������Ϊ�ܽ��Ķ����ȶ��ԡ�
(2) �ܽ��ȶ���������DLVO����
����Ҫ�㣺���������ȶ��Ե�������������������һ����������ķ��»�����һ���ǽ����ʵ���ӽ�ʱ������˫����ص��������ij�����������ȶ�����Σ�ȡ����������������ƽ�⡣�����������ڳ��������������������۳������������������������ȶ����ڡ�
8������ʼ��߷��Ӿۺ�����ܽ�������Ӱ��
(1) ����ʶԾ۳���Ӱ��
��������ʵĴ��ڶ��ܽ����ȶ����ã���������ʵĴ��ڶԽ������ƻ�����(�۳�)��
ʹһ�����ܽ���һ��ʱ������ȫ�۳�������С����ʵ����ʵ���Ũ�ȣ���Ϊ����ʶ��ܽ��ľ۳�ֵ��
�����Ӷ��ܽ��ľ۳�����Ҫ���ã��۳�ֵ�뷴���Ӽ����йء�
һ���������۷����ӵľ۳�ֵ֮�ȴ���Ϊ ���۳�ֵ�뷴���Ӽ�����6�η��ɷ��ȣ�����ղ�һ���Ϲ���
���۳�ֵ�뷴���Ӽ�����6�η��ɷ��ȣ�����ղ�һ���Ϲ���
��������۳����õĻ�����
�� �����Ӽ������ߣ�����ɢ��ĺ��������������ɢ���ص�ʱ�����ij�����
�� ������Ũ�����ߣ������stern�㷴�������࣬�Ӷ����������������ƣ���������ɢ���ص�ʱ�ij�����
(2) �߾�����ӶԾ۳���Ӱ��
���ܽ��м��������߾�����ӿ�ʹ�ܽ��ȶ�(�ǿռ��ȶ����ۼ���ȱ�ȶ�����)���� Ҳ��ʹ�ܽ��۳�����۳��������£�
�� ����ЧӦ���߾������ͨ�������š��ѽ�������һ������۳���
�� ��ˮЧӦ���߾������������ˮ����ˮ�����ýϽ���ˮ������ǿ(��ˮ)���Ӷ��߾���ļ����ȥ������ˮ����ǵı������á�
�� ���к�ЧӦ�������߾���ļ��������ڴ���Ľ������к��˽����ı����ɡ�