一例以急性肾功能不全起病的Caroli综合征伴发ARPKD病例报告并文献复习
杨昊臻1,2,胡瑾华2,童晶晶2,王慧芬2,郑楠楠3
(1、解放军军医进修学院,北京,100853, 2、解放军第302医院肝衰竭诊疗与研究中心,北京,10039,3、解放军第302医院门诊部 北京,10039)
摘要: 目的 探讨Caroli病Ⅱ型合并常染色体隐性遗传多囊肾(ARPKD)的可能机制及诊治方法。方法 回顾性分析我院收治的1例19岁Caroli病Ⅱ型合并ARPKD患者的诊治经过,并复习相关文献。结果 患者以发热伴腹痛起病,分别符合Caroli综合征、ARPKD诊断,彩超示: 双肾增大,皮髓质界线不清,内可见散在多发小囊肿。MRCP:肝内外胆管扩张。胃镜可见食管静脉曲张。结论 Caroli病Ⅱ型可与ARPKD伴发,它们很可能有共同发病机理:PKHD1基因突变。
关键词: Caroli病 Caroli综合征 先天性肝纤维化 常染色体隐性遗传多囊肾 PKHD1基因
A case with acute renal insufficiency of Caroli syndrome comorbid ARPKD and literature review
YANG Hao-zhen,HU Jin-hua,ZHAO Pan,et al(PLA Postgraduate Medical School, Beijing 100853, China ;Liver Failure Treatment and Research Centre,The 302ed Hospital of PLA,Beijing 100039,China)
Abstract: Objective To explore the possible pathogenesis, diagnosis and treatm ent of Caroli typeⅡcomplicated with ARPKD. Methods A 19-year patient diagnosed as Caroli typeⅡcomplicated with ARPKD was identified, and relative documents were reviewed.Results The illness patient was begin with abdominal pain, fever. Ultrasound showed: renal increased in sizes, corticomedullary boundary is not clear,Scattered within multiple small cysts. MRCP: Intrahepatic bile duct and Extrahepatic bile duct dilatation. Gastroscopy showed: esophageal varices. Conclusions Caroli typeⅡshares the similiar pathogenesis with ARPKD (PKHD1gene mutation).
Keywords: Caroli disease;Caroli syndrome;congenital hepatic fibrosis;ARPKD;PKHD1
Caroli病表现为肝内胆管异常扩张,可累及多支,其内可有淤胆及胆石形成,管壁常有有不同程度的纤维化或和急慢性炎症,伴发先天性肝纤维化时称为Caroli病Ⅱ型或Caroli综合征。常染色体隐性遗传多囊肾(Autosemal Recessive Polycystic Kidney disease ARPKD)是一种以多囊肾为表型的常染色体隐性遗传病,多于婴幼儿时期发病,主要特征表现为双肾出现多发的液性囊泡,并进行性长大生长,破坏肾脏的结构和功能,最终导至尿毒症[1]。临床可见Caroli病合并先天性肝纤维化以及ARPKD合并肝纤维化报道,现报道一例Caroli病、先天性肝纤维化、
[1]
1、临床资料
患者,女,19岁,汉族,未婚,因发热、腹痛20天于20##年12月30入院。患者入院前20天无明显诱因出现发热、伴腹痛,最高体温39℃,有乏力、纳差、胸闷。当地予青霉素、左氧氟沙星等抗感染治疗15天,体温有所下降,腹痛稍缓解,停用抗生素后再次出现发热,为进一步诊治来我院。入院查体:生长发育正常,肝区无叩痛,墨菲氏征阴性,腹部压痛、反跳痛阴性,脾左肋下约8cm,质韧,边缘钝,无明显触痛。家族史:其爷爷因“肝硬化”去世,父母及一弟体健,肝肾检查未见异常。患者入院检查血常规:WBC14.83×109/L、N0.85、RBC3.19×1012/L、HGB95g/L、PLT74×109/L;肝功:A/G29/28g/L、Bil17.4/12.7µmol/L、ALT10U/L、AST17U/L、ALP146U/L、GGT68U/L、CHE2121U/L、LDH 176U/L,肾功:Cre476µmol/L、Bun17.6mmol/L;(尿)B2微球蛋白14.45ug/ml、NAG7.4U/L、微量白蛋白91mg/l;24H尿蛋白定量:351.00mg/L。尿常规:白细胞(+++)镜检满视野,蛋白(+),红细胞镜检1-2/HP。大便常规未见异常。甲状腺功能、COOMBS试验、糖水试验、酸溶血试验、肥达、外婓反应(-);RF、抗O正常。CRP207mg/L、ESR72mm/min病毒性肝炎指标阴性。出血热抗体阴性。腹部彩超:1、肝硬化,脾大,少量腹水,2、肝内、外胆管增宽,3、肝多发囊肿,4、门脉高压、侧枝循环开放,5、继发胆囊改变。泌尿系彩超:1、双肾增大,皮髓质界限消失,形态不规则,结构紊乱,内可见多发粟粒样稍强回声。2、双肾轻度局限性积水。3、双肾动脉阻力指数增高。MRCP:1、肝内外胆管扩张,胆总管呈囊肿扩张;2、肝实质弥漫性损害,脾大。3、肝S7囊肿。4、双肾多发囊肿。(如图1、2、3)。胃镜:食管静脉曲张轻度(图4),胆汁反流性胃炎。肝穿结果回报:穿刺组织为纤维瘢痕组织,考虑为肝内胆管性囊肿壁组织,未见肝实质成分。结合临床诊断:1、Caroli病Ⅱ型伴发ARPKD合并1)急性肾盂肾炎 肾功能不全、2)腹水;2、胆汁反流性胃炎。给予补液、抗感染、抑酸及对症治疗后,20##年1月25复查,血常规、肝功正常,肾功:Cre262µmol/L、Bun12.7mmol/L,电解质正常,尿常规正常。
2讨论
2.1临床表现及诊断 本病例一般印象为伴有明显门脉高压的肝肾多发囊性扩张。具体分析如下:
患者青少年,以泌尿系感染发病,伴肾功不全,双肾形态已明显已扩大,皮髓质界限消失,形态不规则,结构紊乱,肾脏弥漫囊肿样表现,加之并发先天性肝纤维化,无阳性家族史,可诊断为ARPKD。虽然Caroli病定义为肝内胆管扩张,但黄志强从外科治疗角度出发,认为Caroli病可合并胆总管囊肿[2],肝内外胆管扩张是否有共同发病机制尚不明了。患者脾大,食管静脉曲张,门脉侧枝循环开放,虽无典型组织学支持,但合并典型先天性肝纤维化表现,故诊断为Caroli病Ⅱ型。患者胆汁反流表现与胆道病变相互关系也是值得进一步探讨的问题,目前已知胆石症胆囊炎患者胆汁反流占48%-79%[3],此类疾病患者多存在胃肌电紊乱及胃排空延迟以及胃肠激素变化。而Caroli病患者胆道病变是否可引起胆汁反流尚无研究。从疾病侧重来看,本患者无反复胆管炎症发作,先天性肝纤维化的门脉高压表现较轻微,ARPKD为疾病主要方面,其继发的反复泌尿系感染和肾功能进行性减退将影响患者预后。
2.2共同发病机制探讨 目前认为ARPKD、CHF、Caroli病与PKHD1(polycystic kidney and hepatic disease1)基因突变相关[4]。该基因定位于人染色体6p21.1-p12。PKHD1在肾脏高度表达,在肝内胆管、胰腺和肺也均有适度表达。PKHD1基因可编码一个由4074个氨基酸组成的单次跨膜受体样蛋白,被称为纤囊素(fibrocystin/ polycystin FPC),此蛋白定位于多种组织器官的细胞上皮原纤毛内[5-7],目前已知肾脏表皮细胞原纤毛可感知流过肾小管的液流并将这种液流引起的机械刺激转化成钙离子信号向细胞内传递,Mayo研究中心的研究也证明肝内胆管上皮细胞原纤毛也具有类似肾脏上皮细胞原纤毛的生理功能:感知胆管内液流,讲将液流引起的机械刺激转化为钙离子信号和cAMP信号,进而影响胆汁分泌[8]。Masyuk TV等人也观察到在小鼠的胆管上皮细胞中使用siRAN阻断纤囊素合成可使原纤毛变短现象[9]。其次纤囊素可作为一个膜受体样蛋白,可将细胞外的信号传递到细胞内,调控体内各管道上皮细胞分化、增殖和移行,从而促进各种生理管道的形成[10]。新近研究表明纤囊素还可能与平面细胞极化(planar cell polarity)的形成过程有关,。在肾脏研究中发现,在正常的肾集合管上皮细胞有丝分裂过程中,细胞纺锤体的方向与集合管的轴向平行,这使集合管上皮细胞沿集合管轴向延伸而不是杂乱无序扩增,而。在PKHD1突变的大鼠模型集合管内,细胞纺锤体不再具有其特定的方向,这种细胞分裂导致管道扩张[11]。笔者假设这种上述机制也存在于肝内,则PKHD1突变导致的干扰平面细胞极化紊乱可形成的机制存在于肝内,影响肝内小胆管细胞排列,使其再塑形障碍,导致先天性肝纤维化发生。籍由此三种疾病便可由共同机制解释:PKHD1突变引起纤囊素表达异常进而导致Caroli病、先天性肝纤维化和ARPKD。
患者下一步治疗将以对症治疗为主,包括预防控制泌尿系感染,避免肾损害加重诱因,肾损害继续进展可考虑血液透析及肾移植治疗;控制门脉高压防止其并发症特别是胃底食管静脉曲张出血,可持续服用降低门脉压药物(需根据肾功能调整剂量);患者肝功基础较好,可耐受手术,通过各种门腔静脉分流手术可有效控制门静脉高压,术后分流型脑病的发生较肝硬化门脉高压患者少。针对Caroli病外科治疗原则是尽可能切除病变组织。肝移植可解除门脉高压,祛除扩张胆管防止其癌变,具有良好的发展前景。
Caroli病、先天性肝纤维化、ARPKD均为少见常染色体隐形遗传病,其三者同时伴发临床报道更为少见。临床工作中若患者出现以其中一方面病变为主的表现应考虑筛查另外两种病变,争取早期发现并发疾病尽早干预。今后试想对其共同发病机制的研究深入,将使其彼此概念内涵与外延将进一步明确,彼此的联系进一步清晰,而其命名或将趋于统一。而针对此类疾病的基因诊断及基因治疗也将早日成为现实。
[参考文献]
[1] Bissler JJ, Dixon BP. Amechanistic approach to inherit edpolycystic kidney disease[J]. Pediatr Nephrol, 2005, 20 (5) :558-561.
[2] 黄志强.Caroli病外科治疗中的问题[J]中华外科杂志,1995,33(11):666-668.
[3] 戴自英,陈灏珠.实用内科学[M].第11版.北京:人民卫生出版社,2001,1743-1744.
[4] Brian R, Nash S, Matthew T. Fibrocystic diseases of the liver [M] Zakim and Boyer's Hepatology: A Textbook of Liver Disease,5th ed. 2006:1374
[5] Xiong H, Chen Y, Yi Y,et al. A novel gene encoding a TIG multipledomain protein is a positional candidate for autosomal recessive polycystic kidney disease[J]Genomics, 2002, 80(1): 96-104.
[6] Christopher J W, Marie C H, Sandro R, et al. The gene mutated in autosomal recessive polycystic kidney disease encodes a large receptor-like protein [J]Nat Genet, 2002, 30(3): 259-269.
 [7] Onuchic L F, Furu L, Nagasawa Y, et al.PKHD1, the polycystic kidney and hepatic disease 1gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet, 2002, 70(5): 1305-1317.
[7] Onuchic L F, Furu L, Nagasawa Y, et al.PKHD1, the polycystic kidney and hepatic disease 1gene, encodes a novel large protein containing multiple immunoglobulin-like plexin-transcription-factor domains and parallel beta-helix 1 repeats. Am J Hum Genet, 2002, 70(5): 1305-1317.
[8] Masyuk TV,Huang BQ, Ward CJ, et al.Defect in cholangiocyte fibrocytin expression and ciliary structure in the PCK rat [J] Gastroenterology. 20## ;125(5):1303-1310.
[9] Masyuk TV, Huang BQ, Masyuk AI, et al.Biliary dysgenesis in the PCK rat, an orthologous model of autosomal recessive polycytic kidney disease. Am [J] Pathol 2004;165:1719-1730.
[10] Kim I, Li C, Liang D, et al. Polycystin-2 expression is regulated by a PC2-binding domain of intracellular portion of fibrocystin[J] BiolChem, 2008, 283(46): 31559~31566.
[11] Fischer E, Legue E, Doyen A et al, Defective planar cell polarity in polycystic kidney disease [J]Nature Genetics, 2006, 38(1) 21-23.
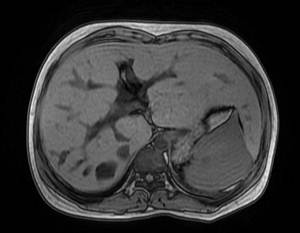
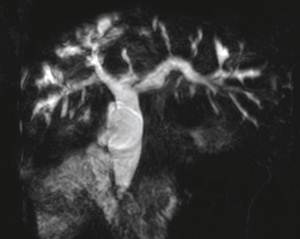
图1、图2 MRCP显示该患者肝内外扩张的胆管
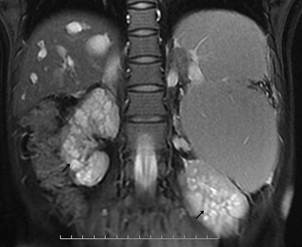
图3显示双肾多发囊肿(黑色尖头↗)
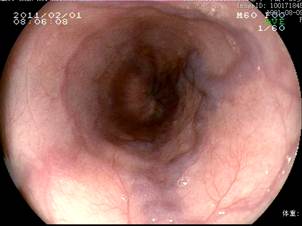
图4胃镜示轻度食管静脉曲张
[1]第一作者简介: 杨昊臻, 军医进修学院20##级硕士研究生, 医师, 研究方向: 肝脏疾病的诊断和治疗,Email:302yhz@gmail.com
通讯作者: 胡瑾华, hjh@ medmail.com.cn
第二篇:Kimura病的临床诊断与治疗一例报告并文献复习
?492?
生堡查速堂盘盍垫!!生!县筮≥!鲞筮!期堡b垫』婪!塑型,』坚!!垫!Q。!垡:!!:塑!:Z
?短篇论著-
Kimura病的临床诊断与治疗一例报告并文献复习
胡涛师晓东刘嵘
Kimura病(Kimuradisease,KD)又称嗜酸性淋巴肉芽肿(eosinophilic
lymphoid
granuloma),是一种良性慢性炎症疾
病,表现为局部肉芽肿病变。本病罕见,临床表现无特异性,易误诊;病程长、易复发,目前无明确统一的治疗原则及有效防止复发的手段。因此,充分认识本病对提高临床诊断水平、正确施治有重要意义。我们报道1例KD患者,并结合文献进行讨论。
病例资料
患儿,男,13岁。因反复颈部淋巴结肿大,伴双眼睑红肿5年人院。肿大淋巴结伴或不伴疼痛,病情可缓解,多次反复。伴反复全身皮疹、瘙痒,多春季出疹,躯干部为著,搔破后结痂,留有色素脱失斑。生长发育及入学正常。无类似家族史。查体:全身散在粟粒大小色素脱失斑,双下肢散在丘疹样皮疹,部分破溃结痂,躯干散在数个皮下结节,1—3
cm,
质韧,无压痛;双眼睑红肿;颈部、耳后、颌下、颏下淋巴结肿大,部分发生融合;肝脾无肿大。血常规:WBC
19.63×
109/L,嗜酸粒细胞0.398,淋巴细胞0.238,中性粒细胞
0.339,Hb120
g/L,BPC291×109/L。尿常规、血尿素氮及肌
酐正常。ALT、&ST及心肌酶正常。铁蛋白140峭/L。,I'B抗
体及PPD阴性。纤维蛋白降解产物≥20me/L;D?二聚体
1523∥L。微丝蚴及丝虫阴性,肝吸虫及血吸虫抗体阴性。
Igg>80kU/L,IgU29.09
g/L,IgA
3.25
g/L,IgG21.37g/L。
淋巴细胞亚群:CD3+细胞为74%,CDl9+细胞为14%,CD3+CD4+细胞为48%,CD3+CD8+细胞为26%,CD4+/CD8+细胞比值1.86。骨髓增生活跃,粒:红为4.1:1;粒系增生,部分粒细胞有空泡;中幼(占0.13)、晚幼(占0.13)、杆状核嗜酸粒细胞(占0.02)均增高,分叶核嗜酸粒细胞正常;红细胞、淋巴细胞、单核细胞、巨核细胞系及血小板无异常。B超:双侧腮腺、颌下及颈部淋巴结肿大;双眼球无异常。CT显示双侧泪腺炎性假瘤。胸部增强CT:双肺纹理增多,颏下、锁骨下、腋窝淋巴结肿大,肝内多个点状高密度影。过敏原:豚草属、柳属、草类混合物及蚊子呈阳性。左锁骨上淋巴结活检显示小血管增生明显,淋巴细胞丰富,形成淋巴滤泡,滤泡内有生发中心,生发中心大量血管形成;嗜酸粒细胞浸润明显,部分区域保持淋巴结正常结构,病理诊断KD病。予地塞米松15ng/d×7d,病情迅速缓解,后予以泼尼松121服并渐减
DOI:10.3760/ema.j.iasn.0253-2727.2010.07.018
作者单位:100020北京,首都儿科研究所附属儿童医院血液科
通信作者:师晓东,Email:蛐usan28@鲫ail.P.om
万方数据
量,病情稳定出院。
讨论及文献复习
KD首先由我国金显宅等1937年报道,以嗜伊红细胞性增生性淋巴母细胞瘤命名,曾称为金氏病(Kimm氏病)。1948年,木村哲二等描述为一种不寻常的肉芽组织伴淋巴组织增生,称为木村病(Kimura病)。多见于东亚地区亚裔青
壮年男性,好发年龄20~39岁(中位年龄31.5岁),男女比例3.5—6.O:l¨…。全球报道300余例嵋1,非亚裔人口也有散发,包括白种人、西班牙人、阿拉伯人以及黑种人【3J。
1.病因及发病机制:尚不明确,多数学者认为KD是一种免疫功能紊乱导致的慢性炎症性疾病。迄今发现多种致病因素,包括自身免疫功能异常、过敏、肿瘤、蚊虫叮咬、接触猫科动物、与寄生虫和白色念珠菌有关的免疫性疾病等”…。部分患者伴哮喘、湿疹、过敏性皮炎、过敏性紫癜、免疫性血小板减少症、免疫性溶血性贫血、肾病、溃疡性结肠炎等变态反应性疾病忙1。l例KD患者也可同时合并多种变态反应性疾病∽o。肾移植患者在慢性移植排斥反应后也可发生KD¨J。既往认为Rubinstein.Taybi综合征(RTS,阔拇指巨趾综合征)伴有免疫缺陷,目前认为是伴有免疫功能异常的先天性疾病,最近有RTS患者伴KD的报道¨1。上述报道均支持免疫功能异常可能导致KD,并提示任何导致免疫功能异常的原因均可能成为KD的病因。
KD患者外周血嗜酸粒细胞、肥大细胞及IgE增高¨J,I型变态反应也有上述变化;KD患者Th2细胞、IL-4、IL-5、IL-6、IL-13升高”,1,IL4、II,-5是主要的Th2细胞因子,与B细胞增殖、成熟及促抗体生成有关。IL4在诱导B细胞产生IgE过程中至关重要,而IL-5可激活嗜酸粒细胞,导致炎症反应。因此Th2细胞及其细胞因子在KD发病中扮演重要角色。这些研究从细胞水平支持变态反应可能是KD的发病机制。
目前尚无KD家系报道,缺乏基因水平的研究报告,尚无证据本病与遗传有关。
2.临床表现:本病罕见,临床表现缺乏特异性,易误诊。主要表现为软组织结节及淋巴结肿大,病灶单发或多发,病变可累及全身,以头颈部最常见,多为深部皮下肿块,大的可达3~10cm,可累及整个区域的淋巴结及唾液腺。即使累及耳廓也是软组织病变,无软骨受累¨引。1例KD患者以呼吸
吞咽困难就诊,伴吸气性喉鸣,病变为会厌部软组织肿块oIll。1例病史已3年的KD患者,因左上肢无力、视物不清复诊,MPd显示右颈内动脉闭塞,右侧椎动脉及左锁骨下
生堡查邃堂盘查垫!垒生Z旦筮i!鲞筮!魍£!垫』堕型,』!k垫!旦:型:!!:丛:!
?493?
动脉起始部狭窄,右侧颞顶叶脑梗死,经激素及阿司匹林治疗后恢复¨“。眼睑、口腔黏膜、舌等少见部位病变也有报道。有些病例仅表现为浅表淋巴结肿大,包括颈部、腋窝、滑车上及腹股沟淋巴结。无深部淋巴结及脏器受累¨“。骨骼受累以扁平骨较多,长骨少见,可累及关节,伴骨膜反应”“。皮肤受累表现为不规则红斑、色素沉着、脱屑、变厚、粗糙等,伴瘙痒¨31,也有合并疥癣、风疹、湿疹的报道。此外输精管和神经等罕见部位也有报道¨“。
KD多为局部病理进程,无系统症状或体征,但可累及脏器,肾脏最常见,表现为肾病综合征(NS),Ns可先于肿块发生¨“。甚至l例患者3岁时诊断为NS,10年后出现嗜酸粒细胞增高和EPO不敏感性贫血,激素治疗后恢复,最终诊断KD¨引。肝、脾也可受累,表现为脏器内团块,颇似恶性肿瘤转移。少数病例可有全身症状,如发热、盗汗、乏力、顽固性高血压等;还可能引起类“高嗜酸粒细胞综合征”表现,如刺激性干咳等。
本例患儿泪腺受累。既往未见报道。累及眼眶及眼附属器官的疾病表现为突眼、眼睑肿胀、可能影响视力,应注意与绿色瘤鉴别。
3.实验室检查:KD患者外周血嗜酸粒细胞及IgE增高,嗜酸粒细胞比例多在0.20左右。有时可达0.60—0.70,个别患者嗜酸粒细胞不高。嗜酸粒细胞和IgE水平可作为病情活动程度、疗效观察、评估预后的指标。患者TId细胞、肥大细胞、TNF—01、IL4、IL-5、1I..-13增高。应注意脏器功能或影像学检查,判断病变范围。过敏原检查和机体免疫功能检测有助于发现病因或诱因。
文献报道的病例均有骨髓嗜酸粒细胞增高,本例患儿骨髓嗜酸粒细胞增高,分类结果为中幼粒、晚幼粒及杆状核嗜酸粒细胞比例增高,分叶核嗜酸粒细胞比例正常,原始及早幼粒细胞比例正常,提示骨髓嗜酸粒细胞系增生活跃,无成熟障碍,更不同于慢性嗜酸粒细胞自血病等血液肿瘤性疾病。
4.影像学检查:影像学检查缺乏特异性,累及软组织时界限不清,易误诊为恶性肿瘤。CT对KD无特异诊断意义。MRI检查r12相高信号,Tl相低或中等信号,但也不尽然,与病变部位纤维化及血管增生程度有关【17J。B超显示软组织病变中心低回声,外周高回声;肿胀的淋巴结为圆形实体,轮廓清晰,淋巴结内为均匀的高回声,可以清晰地看到血管进入淋巴结;由于血管血供丰富,易与恶性肿瘤混淆,但恶性肿瘤为血流高阻力,KD则比正常组织血流阻力低…J。无论如何,影像检查仅可作为了解病变部位、血管情况、与周围组织关系的手段,确诊还需依靠病理学。
5.病理学检查:病变很少累及真皮,多累及深部软组织,可深达横纹肌和神经外膜¨…。不同部位的KD基本病理表现大致相同。淋巴细胞、嗜酸粒细胞、小血管和纤维组织以不同比例分布,小血管增生明显,血管内皮细胞呈扁平或立方状,胞质内无空泡,管壁较薄;淋巴细胞丰富,形成大小不一的淋巴滤泡,滤泡内有活跃的生发中心,并有大量血管
万方数据
形成,可见多核细胞、蛋白样物沉积及坏死;嗜酸粒细胞浸润,多有嗜酸粒细胞微脓肿;纤维化、硬化也是KD的典型特点。硬化区域炎性细胞减少,血管萎缩。累及淋巴结时,病理表现与软组织类似,部分区域仍有淋巴结正常结构【I“。免疫学表型符合淋巴组织的表型,病变组织的淋巴滤泡表达B细胞抗原,滤泡间淋巴细胞多表达T细胞标志¨…。肾损害时,肾间质嗜酸粒细胞浸润是主要特征Ⅲj,病理可为肾小球系膜增生病变、膜性病变、微小病变、局灶节段性肾小球硬
化、弥漫增生性病变、IgA肾病、IgM肾病等‘猢“,通过肾活检
不能诊断KD。目前尚无皮损病理检查报道。
6.诊断:皮下软组织肿块及淋巴结肿大,嗜酸粒细胞及IgE增高,亚裔患者应考虑KD的可能一J,通过病理检查可确诊,有时病理学可能是诊断的唯一依据。影像学、脏器功能、过敏原检测有助于全面评估病情,对治疗方案的选择有指导意义。
7.鉴别诊断:注意和淋巴瘤、涎腺肿瘤、淋巴结炎、淋巴结结核、嗜酸粒细胞肉芽肿、血管肿瘤、Kaposi肉瘤、Castle—m册病、脓性肉芽肿等疾病鉴别,这些疾病多被临床医生认识。当KD被了解后,通过病理鉴别并不困难。
病理诊断时应注意与血管淋巴组织增生伴嗜酸性粒细胞浸润(ALHE)鉴别,ALHE多为头颈部软组织肿块,易复发,病理可见淋巴细胞及嗜酸粒细胞浸润、血管增生,但ALHE多见于西方中年女性,外周血嗜酸粒细胞多低于0.10,IsE不高,病理检查有或无淋巴滤泡,少有活跃的生发中心,免疫表型复合血管瘤表型¨8’19J。
应注意本病与高嗜酸粒细胞综合征(HES)的区别,HES临床表现不以软组织包块为主,诊断要求未发现引起嗜酸粒细胞增多的常见原因,由于嗜酸粒细胞的细胞毒作用,引起器官损害,常见心脏、神经系统、呼吸道及皮肤受累,最终导致血栓形成及纤维化。特发性高嗜酸粒细胞综合征(IHES)是HES的分型之一,没有任何克隆性细胞遗传学异常的HES应考虑IHESt圳。
8.预后:KD病程长,多呈进展性,主要问题是易复发。外周血嗜酸粒细胞>0.50、IgE>10
000
kU/L、多发性唾液腺
外病变是易复发的预后因素一J。目前尚无KD直接致死的报道。但累及会厌、重要血管闭塞时,也可能危及生命。
9.治疗:常用的治疗方法包括手术、放疗、激素。病变部位局限时首选手术,由于和周围组织界限不清,切除不完全时易复发。Ussmuller等∞1主张切除肿块要彻底,应附带一些周边健康组织,切除淋巴结肿块可能减低肾病复发率,达到长期缓解。但多发病灶、病变范围广、脏器内多发结节等情况时手术受限。局部病变也可放疗,KD对放疗敏感,放疗后血管和内皮细胞形态恢复正常、嗜酸粒细胞消失、淋巴细胞浸润和滤泡减少、纤维成分增多。放疗适用于局部病变、不宜手术或术前需缩小肿块者;术后放疗可降低复发率,推荐小剂量放疗,总剂量26~30cy,放疗后复发再次放疗仍有效¨L…。放疗可致第二肿瘤,影响生长发育及生育功能,而KD多数预后良好,因此我们认为病变范围广泛或儿童患
?,194?
生垡查速堂苤盍!Q!Q生!旦笠j!鲞筮!翅鱼h也』丛!塑坐尘:!坠垃垫!Q:!尘:!!:堕!:!
者不应首选放疗。激素起效快,效果明显,合并有哮喘、肾病、自身免疫性溶血等情况时可首选,停药也易复发。此外,还有化疗及细胞毒性药物治疗KD的报道,如CHOP方案或环磷酰胺、长春新碱等,主要用于激素耐药者ⅢJ。由于上述治疗不良反应大和(或)易复发,有学者认为初诊可不积极治疗,当出现明显症状或功能受累时再治疗。
随着对病因学和发病机制的研究,近年出现一些针对免疫状态的新型疗法。手术或放疗后应用环孢素(3~5mg?l【g~?d“),可维持嗜酸粒细胞、IL-4、IL-5、IL一13水平在正常范围,减少复发,也可用于复发病例Ⅲ】。伊马替尼选择性抑制蛋白酪氨酸激酶,在治疗HES中可发挥作用,因此认为也可治疗KD拉o。Pranlukast(普仑司特)为白三烯受体拮抗剂(LTRAs),减少IL.4的生成,用于治疗过敏性疾病,IL4增高是KD重要的发病机制,故可使用,有报道应用Pranlukast可控制KD的肿块生长和过敏症状,剂量
450
mg/d,治疗期间无明显不良反应∞1。此外也有全反式维
甲酸、己酮可可碱、甲磺司特、激光等试验性治疗的报道ⅢJ。
综上所述,KD是一种全身性系统性疾病,各器官系统均可能受累,我们认为KD患者机体免疫功能紊乱,属于一种变态反应性疾病。由于KD是慢性疾病、病程长,几乎无致死病例报道,故确诊后可不急于治疗。首先应确定病变范围、有无脏器受累及合并症,了解机体免疫状态,积极寻找病因或诱因(如过敏原等)。全面评估病情是合理选择治疗方案的基础。病灶局限无合并症者可采用手术和(或)放疗;儿童期或病变范围广者不宜手术或放疗;合并自身免疫性疾病时可选用激素或免疫抑制剂;病情稳定、无急需处理者,可尝试不良反应小的新型治疗,如蛋白酪氨酸激酶抑制剂、白三烯受体拮抗剂、环孢素等。由于本病与过敏性疾病密切相关,过敏多为诱因,病程迁延反复且明确过敏原时,可考虑抗过敏或脱敏治疗,无效时再按上述原则治疗。
参考文献
[1]Takeishi
M,MakinaY,NishiokaH,eta1.Kimuradisease:diag-
nostie
imaging
findingsands.rocaltrealmcnt.JCraniofaeSurg,
2007.18:1062-1067.
[2]SunQF,xuDZ,PansH,etal.Kimuradisease:reviewoftllelit-
eralure.IntemMedJ,2008,38:668-674.
[3]ChenH,ThompsonLDR,AguileraNSI,eta1.Kimuradisease,a
elinieopathologiestudyof21
cas∞.AmJStagPathol,2004,28:
505-513.
[4]杨弋超.Kimura病的诊断与治疗.皖南医学院学报,2008,27:
97.
[5]OhtsukaY,Shimim
T,Fujii
T,eta1.Pranlukastm殍lllIgt档turnout
growthby
attenuatingII,4
production
in
Kimuradisease.
EurJPediatr,2004,163:416-417.
[6]MehulPD,KatherineMS,Erika
B,et
a1.Kimuradiseasewithad-
vancedrenal
damage
with
anti—tubularbasementmembraneanti-
body.PediatrNephrol,2004,19:1404-1407.
万方数据
[7]h
HJ,TsaiJD,SheuJC,cta1.Kimura
diseasein
apatient
with
renal
allog础tfailuresecondarytochronicrejection.PediatrNeph-
ml。2003,18:1069-1072.
[8]KimCJ,NamJH,Chung
HY,et
a1.Kimuradiseasein
a
patient
withRubinstein-Taybisyndrome.PediatrInt,2004,46:609-611.
[9]1wai
H,NakaeK,IkedaK,et
a1.Kimuradisease:diagnosisand
prognosticfactors.OtolaryngolHead
NeckStag,2007,137:306?
311.
[10]林尚泽.耳廓木村(Kimura病).国外医学耳鼻咽喉科学分册,2006,26:168—169.
[11]
BadrA,HallemAA,CarlsenEd.Kimuradiseaseofepiglottis.
HeadNeckPath01.2008.2:328-332.
[12]付胜奇,邢红霞,张在强,等.Kimura病致脑动脉狭窄伴梗死并文献复习.临床误诊误治。2008,21:90-91.
[13]姜蕾,姜晓钟,赵云富,等.头颈部Kimura病的临床病理特点与诊断.临床与实验病理学杂志,2007,23:321-323.
[14]Varshney
MK,Kumar
A,KhanSA,eta1.Kimumdiseaseof
ex-
tremity:Unusualmanifestationin
a
longbone.Joint
Bone
Spine,
2008.75:492-494.
[15]周康,郝玉书.Kimum病.中华血液学杂志,2005,26:253-255.
[16]HungCC。LiaoPL,ChangCT,eta1.Steroid?sensitiveanelIlia
in
a
boy
on
dialysisan
associationwithKimuradisezme.PMiatr
Nephrel,2000。15:183-185.
[17]杨志云,赖英荣。冯崇锦,等.颌面部Kimura病CT和MR诊断.I临床放射学杂志,2006,25:1016—1018.
[18]常晓燕,陈杰.血管淋巴组织增生伴嗜酸性粒细胞浸润与
kimura病的鉴别诊断.诊断病理学杂志,2004,1
1:l19-121.
[19]陆磊,陈仁贵,李小秋.Kimura病和上皮样血管瘤的临床病理学观察.中华病理学杂志,2005.34:353—357.
[20]刘春蓓.胡伟新,陈惠萍,等.Kimura病的肾脏损害.肾脏病与透析肾移植杂志,2004,13:524-529.
[21]DedeF,AyhD,AtilganKG,eta1.Focalsegmentalglomerulo-
sclerosisassociatingkimuradisease.RenalFailure,2005,27:
353-355.
[22]
沈莉菁,韩洁英,陈芳源.原发性嗜酸粒细胞增多症的诊疗进展.临床血液学杂志,2008,21:285-288.
[23]u∞muuerJ,DonatHK,Shimizu
M,et
a1.Differentialdiagnosis
oftumorous
space-occupying
lesionsoftheparotidgland:angi-
olymphoidhyperplasiawitheosinophiliaandKimuradisease.La-
ryngorhinootologie,1997。76:110-115.
[24]DayTA,AbreoF,Ho由¥ocDK,eta1.TreatmentofKimttradi铲
P-,aSC:a
therapeuticenigma.OtolaryngolHeadNeckSurg,1995,
1
12:333-337.
[25]林沁,庄永泽,陈淑娇.Kimura病l例附文献复习.实用医学杂志,2006,22:2284-2286.
[26]
王友元,黄志权,陈伟良,等.多发性Kimura病I例报道及文
献复习.中国121腔颌面外科杂志,2007,9:392-395.
(收稿日期:2010-01-08)

Kimura病的临床诊断与治疗一例报告并文献复习
作者:
作者单位:
刊名:
英文刊名:
年,卷(期):胡涛, 师晓东, 刘嵘首都儿科研究所附属儿童医院血液科,北京,100020中华血液学杂志CHINESE JOURNAL OF HEMATOLOGY2010,31(7)
参考文献(26条)
1.Iwai H;Nakae K;Ikeda K Kimura disease; diagnosis and prognostic factors 2007
2.Kim CJ;Nam JH;Chung HY Kimura disease in a patientwith Rubinstein-Taybi syndrome 2004
3.Lu HJ;Tsai JD;Sheu JC Kimura disease in a patient with renal allograft failure secondary tochronic rejection 2003
4.Mehul PD;Katherine MS;Erika B Kimura disease with advanced renal damage with anti-tubular basementmembrane antibody 2004
5.Ohtsuka Y;Shimizu T;Fujii T Pranlukast regulagtes tumour growth by attenuating IL-4 production inKimura disease 2004
6.Hung CC;Liao PL;Chang CT Steroid-sensitive anemia in a boy on dialysisan association with Kimuradisease[外文期刊] 2000
7.周康;郝玉书 Kimura病[期刊论文]-中华血液学杂志 2005(4)
8.Varshney MK;Kumar A;Khan SA Kimura disease of extremity; Unusual manifestation in a long bone 2008
9.姜蕾;姜晓钟;赵云富 头颈部Kimura病的临床病理特点与诊断 2007
10.付胜奇;邢红霞;张在强 Kimura病致脑动脉狭窄伴梗死并文献复习[期刊论文]-临床误诊误治 2008(4)
11.杨弋超 Kimura病的诊断与治疗 2008
12.Chen H;Thompson LDR;Aguilera NSI Kimura disease,a clinicopathologic study of 21 cases 2004
13.Sun QF;Xu DZ;Pan SH Kimura disease:review of the literature 2008
14.王友元;黄志权;陈伟良 多发性Kimura病1例报道及文献复习[期刊论文]-中国口腔颌面外科杂志 2007(5)
15.林沁;庄永泽;陈淑娇 Kimura病1例附文献复习[期刊论文]-实用医学杂志 2006(19)
16.Day TA;Abreo F;Hoajsoc DK Treatment of Kimura disease:atherapeutic enigma 1995
17.Ussmuller J;Donat HK;Shimizu M Differential diagnosis of tumorous space-occupying lesions of theparotid gland; angi-olymphoid hyperplasia with eosinophilia and Kimura disease 1997
18.沈莉菁;韩洁英;陈芳源 原发性嗜酸粒细胞增多症的诊疗进展[期刊论文]-临床血液学杂志 2008(3)
19.Dede F;Ayh D;Atilgan KG Focal segmental glomerulo-sclerosis associating kimura disease 2005
20.刘春蓓;胡伟新;陈惠萍 Kimura病的肾脏损害[期刊论文]-肾脏病与透析肾移植杂志 2004(6)
21.陆磊;陈仁贵;李小秋 Kimura病和上皮样血管瘤的临床病理学观察[期刊论文]-中华病理学杂志 2005(6)
22.常晓燕;陈杰 血管淋巴组织增生伴嗜酸性粒细胞浸润与kimum病的鉴别诊断 2004
23.杨志云;赖英荣;冯崇锦 颌面部Kimura病CT和MR诊断 2006
24.Badr A;Hallem AA;Carlsen Ed Kimura disease of epiglottis 2008
25.林尚泽 耳廓木村(Kimura病) 2006
26.Takeishi M;Makina Y;Nishioka H Kimura disease:diagnostic imaging findings and surgical treatment2007
本文链接:http://d..cn/Periodical_zhxyx201007016.aspx