小学期药物化学实验
学院: 药学院
分组: 一组
姓名:
学号:
指导老师:
贝诺酯的合成
摘要:本品以阿司匹林和扑热息痛(对乙酰氨基苯酚)为原料合成。阿司匹林与氯化亚砜反应制备乙酰水杨酰氯时以少量吡啶作催化剂,氯化亚砜过量20%左右以提高收率,过量氯化亚砜减压蒸馏除去。本品经口服进入体内后,经酯酶作用,释放出阿司匹林和扑热息痛而产年药效。本品既有阿司匹林的解热镇痛抗炎作用,又保持了扑热息痛的解热作用。
关键词 贝诺酯;溶剂;合成条件
一、实验目的
1. 通过乙酰水杨酰氯的制备,了解氯化试剂的选择及操作中的注意事项。
2. 通过本实验了解酯化反应原理,掌握无水操作的技能及反应中产生有害气体的吸收方法。
二、实验原理
扑炎痛为消炎镇痛药,常以阿司匹林和扑热息痛(对乙酰氨基苯酚)为原料合成。阿司匹林与氯化亚砜反应制备乙酰水杨酰氯时以少量吡啶作催化剂,氯化亚砜过量20%左右以提高收率,过量氯化亚砜减压蒸馏除去。经过上述合成,扑炎痛既保留了解热镇痛的作用,又减小了原药的毒副作用,并有协同作用,适用于急慢性风湿性关节炎、风湿痛、头痛及感冒发烧等病症的治疗。合成反应如下:

三、实验仪器
1 原料与试剂:阿司匹林(药用)、扑热息痛(化学纯)、二氯亚砜(分析纯)、吡啶(分析纯)、氢氧化钠(化学纯)、苯(化学纯)、甲苯(化学纯)、二甲苯(化学纯)、丙酮(化学纯)
2 主要仪器:搅拌机、电热套、铁架台、恒温水浴锅、三口瓶(150ml)、直形冷凝管、抽滤瓶(250ml)、布氏漏斗(60mm)、温度计(100℃)、量筒、滴管
表1-物料规格及配比
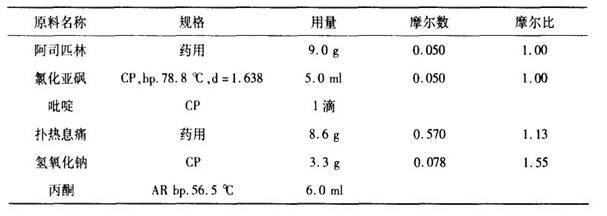
表2-物理性质
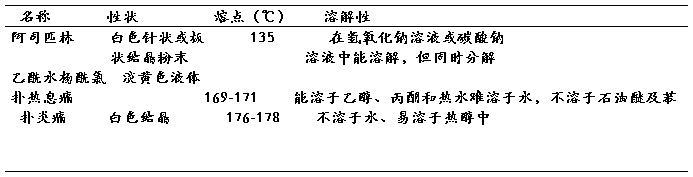
四、实验内容
1.实验操作
(1)乙酰水杨酰氯的制备
在装有回流冷凝器(上端附有氯化钙干燥管、排气导管通入氢氧化钠溶液吸收)、温度计的150 ml三颈瓶中,加入止爆剂、阿司匹林9 g,氯化亚砜5 m1,滴入吡啶1滴(催化反应用),缓缓加热至75℃,维持70—75℃,保温反应至无气体逸出(约2~3 h)。反应完结改成减压蒸馏装置,减压蒸除过量的氯化亚砜后(防止倒吸),冷却,得乙酰水杨酰氯,加入无水丙酮6 ml,混匀密封备用。
(2)扑炎痛的制备
另在装有搅拌、恒压滴液漏斗、温度计的150 ml三颈瓶中,加入扑热息痛8.6g,水50 ml,搅拌下,于10~15℃缓缓加入氢氧化钠液18 ml(氢氧化钠3.3 g加水至18 m1),降温至8~12℃,慢慢滴加上述制得的乙酰水杨酰氯无水丙酮液(约20 min滴毕),调节pH9~10,于20~25℃搅拌1.5~2 h。反应毕,抽滤,用水洗至中性,烘干,得粗品。
(3)精制扑炎痛
将粗品用95%乙醇(样品:95%乙醇=1:8)精制,约得精品5~7 g,mp.174~178℃.收率为44%.具体方法为:
取粗品5 g置于装有球形冷凝器的100 ml圆底瓶中,加入10倍量(w/v)95% 乙醇,在水浴上加热溶解。稍冷,加活性碳脱色(活性碳用量视粗品颜色而定),加热回流30 min,趁热抽滤(布氏漏斗、抽滤瓶应预热)。将滤液趁热转移至烧杯中,自然冷却,待结晶完全析出后,抽滤,压干;用少量乙醇洗涤两次(母液回收),压干,干燥,测熔点,计算收率。
五、实验结果
成品的鉴别
1、取少量成品和扑热息痛原料分别滴加氯化铁溶液,扑热息痛加入氯化铁溶液会显蓝紫色,成品加入氯化铁溶液后无显色反应,说明所得成品中不含有扑热息痛的原料杂质。
2、用熔点测定仪微量法分别测原料和成品的熔点,测得成品熔点为169~174℃,阿司匹林135℃,贝诺酯174℃,扑热息痛169-171℃,与文献报道一致,说明所得成品中不含有阿司匹林的原料杂质。
3、采用薄层检验方法定性判断成品中的所含成分,使用的薄层展开剂:乙酸乙酯-乙醚-冰醋酸(12:7:1),测得结果为成品中不含有原料扑热息痛和阿司匹林的成分。
六、实验讨论及分析
粗制的过程中,首先,由于仪器和人为因素的影响,没有控制在在110-115℃的温度范围之内,因为用的是电热套、油浴锅,也不能保持恒温,不小心温度也有骤升骤降的情况,导致超出适宜反应温度,这样的情况使产率降低,同时副产物增多。精制后,用TLC进行定性检测,发现抽滤得到的滤液和反应瓶中残留的黑色固体均有对乙酰氨基酚,导致产率低,原因是浓缩过度或没有选择合适的溶剂将残留的固体溶解提取,导致产物的流失。
参考文献
[1] 王文静,吕玮,卢泽.贝诺酯的合成[J].河南大学学报:医学版,2006,25(1):39—42.
[2]韦正友,郭荷民,黄勤安主编.医学有机化学实验教程.安徽科学技术出版社,2007.1. p101页
[3]刘抚梅. 药物化学[M]. 北京:中国医药科技出版社,2003:376-378
第二篇:20xx级药物化学实验
药物化学实验
实验带教:
药学一组:徐启贵 136xxxxxxxx
药学二组:梁国娟 139xxxxxxxx
药学三组:徐启贵
药物分析:徐启贵
工业制药:胡湘南 66729212
实验准备:徐启贵
一、实验要求
1. 药物化学实验是对学生进行综合训练,除了巩固学生的基本操作技术和技能,要求培养学生正确选择有机反应装置,认真分析反应过程的现象和影响因素,熟练产物的分离提纯鉴定。通过这个教学环节,较全面培养学生的动手能力和学会分析问题和解决问题,为今后学习专业课和开展科研奠定良好的基础。
2. 进行药物化学实验前,应该复习有机化学实验中所涉及的基本操作,原则上应该带有机化学实验讲义参加药物化学实验。药物化学课程一般不单独对实验基本操作技术进行讲授和示范。
3. 学生进行实验前,应对相关的有机化合物的性能有所了解,因而本课要求学生学会使用有关的手册、文献资料及信息网络查阅有机化合物的物理化学常数。
4. 要求学生实验前认真预习,写出预习笔记;实验课中仔细观察和正确记录,整理分析数据;课后按规范书写实验报告。
5. 安全、卫生、节约药品的教育和实施贯穿始终。
6. 实验室轮流值日,值日生必须在全部实验结束后经老师同意方可离开!!!
二、实验报告
包括实验目的,实验方案步骤,实验结果并讨论分析在实验中出现的问题,实验中的特殊现象、实验操作的成败、实验的关键点等内容进行整理、解释、分析总结,提出实验结论或提出自己的看法等。
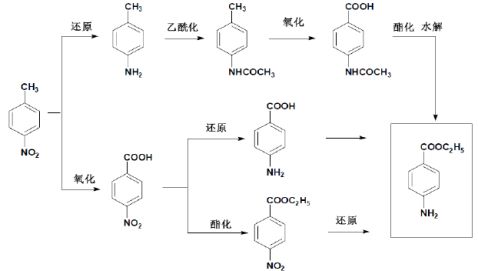
实验一:苯佐卡因的合成(必做实验)
[反应式
]
[试剂]
对硝基甲苯,重铬酸钾,浓硫酸,浓盐酸,氢氧化钠,锡粉,氨水,冰醋酸,无水乙醇,碳酸钠,95% 乙醇,活性碳
[步骤]
1. 对硝基苯甲酸的制备(氧化)
在磁力搅拌器上[1],在装有磁力搅拌子和球型冷凝器的250 m1三颈瓶中,加入重铬酸钾24.0 g,水50 m1,开动搅拌,待重铬酸钾溶解后,加入研碎的对硝基甲苯8.0 g,用滴液漏斗[2]滴加32 m1浓硫酸,严防反应混合物高于沸腾温度,滴加时间约20~30 min [3]。随着浓硫酸的加入,氧化反应随之开始,反应温度迅速上升,料液颜色逐渐变深。滴加完毕,加热,保持反应液微沸60~90 min [4]。停止加热。冷却后,将反应液慢慢倾入80 m1冷水中,抽滤,固体用45 m1水分三次洗涤后,转移到烧杯中,加入5% 硫酸35 m1,在沸水浴上加热10 min,并不时搅拌,冷却后抽滤,将固体溶于温热[5]的5%氢氧化钠溶液70 m1中,在50℃左右抽滤[6],滤液加入活性碳0.5 g脱色(5~10 min),趁热抽滤。冷却,在充分搅拌下,将滤液慢慢倒入15% 硫酸50 m1中,浅黄色沉淀立即析出。用试纸检验溶液是否呈酸性。呈酸性后抽滤,固体用少量水洗至中性,抽干后放置晾干,而后称重。必要时再用50%乙醇重结晶[7],
可得到浅黄色小针状晶体。产量:约4 g。纯对硝基苯甲酸为浅黄色单斜叶片状晶体,熔点242℃。
2. 对氨基苯甲酸的制备(还原)[8]
称取4 g对硝基苯甲酸、9 g锡粉加入到放有磁力搅拌子的100m1圆底烧瓶中,固定在磁力搅拌器上,装上回流冷凝管,开动搅拌,从冷凝管上口分批加入20 m1浓盐酸[9],反应立即开始(必要时可用微热片刻以保持反应正常进行),反应液中锡粉逐渐减少,当反应接近终点时(约20~30min),反应液呈透明状。稍冷,将反应液倾倒入250 m1 烧杯中,用少量水洗涤留存的锡块固体。反应液冷至室温,慢慢地滴加浓氨水,边滴加边搅拌,直至溶液对pH纸刚好呈碱性。滤去析出的氢氧化锡沉淀[10],沉淀用少许水洗涤,合并滤液和洗液。注意总体积不要超过55 m1[11],若总体积超过55 m1,在水浴上加热浓缩至45~55m1,浓缩过程中若有固体析出,应滤去。向滤液中小心地滴加冰醋酸[12],至对蓝色石蕊试纸恰好呈酸性乃有白色晶体析出,再滴加少量冰醋酸至溶液pH 6-7,有更多的固体析出。在冷水浴中冷却,滤集产品[13]得白色固体,在空气中晾干称重。产量:约2 g。纯对氨基苯甲酸为白色絮状晶体,于186℃熔融并分解。
3. 对氨基苯甲酸乙酯(酯化)[14]
将2 g对氨基苯甲酸,20 m1无水乙醇,2.5 m1浓硫酸[15]放入100m1 圆底烧瓶中,混匀后投入沸石,水浴加热回流1~1.5 h,反应液呈无色透明状。将反应液趁热[16]倒入装有85 m1冷水的250 m1烧杯中,得一透明溶液。溶液稍冷后,慢慢加入碳酸钠固体粉末[17],边加边搅拌,使碳酸钠粉末充分溶解,当液面有少许白色沉淀出现时,慢慢加入10%碳酸钠溶液,将溶液pH调至呈中性,滤集沉淀,少量水洗涤,抽干,空气中晾干后称重。必要时可用50%乙醇重结晶。产量:1~2 g。纯对氨基苯甲酸乙酯为白色针状晶体,熔点92℃。
4. 精制
将粗品置于装有球形冷凝器的100 m1 圆底瓶中,加入10~15 倍(m1/g)50%乙醇,在水浴上加热溶解。稍冷,加活性碳脱色(活性碳用量视粗品颜色而定),加热回流20 min,趁热抽滤(布氏漏斗、抽滤瓶应预热)。将滤液趁热转移至烧杯中,自然冷却,待结晶完全析出后,抽滤,用少量50%乙醇洗涤两次,压干,干燥,测熔点,计算收率。
[注释]
[1]本氧化反应十分激烈。采用有效搅拌和滴加硫酸的方法可使反应较平稳、安全。装置安装完毕后应经教师检查无误后再加料使用。
[2]滴液漏斗在使用前要检查其密封性是否完好。
[3]若滴加硫酸时烧瓶内有较多白色烟雾或火花出现,则应迅速减慢或暂停滴加,必要时用冷水浴冷却烧瓶。
[4]反应温度过高,一部分对硝基苯甲酸会挥发,冷结于冷凝管内壁上。此时可适当关小冷凝水,让其熔融滴入。
[5]反应式为:Cr2(SO4)3 + 6NaOH 3 + 3Na2SO4 而Cr(OH)3是两性物质,在温度较高时又会溶于碱中:2Cr(OH)3 + NaOH 2+2H2O,故加热溶解时温度须在60℃以下。
[6]氧化反应一步在用5%氢氧化钠处理滤渣时,温度应保持在50℃左右,若温度过低,对硝基苯甲酸钠会析出而被滤去。
[7]本产品也可用升华法精制。
[8]还原反应中加料次序不要颠倒,加热时用小火。
[9]还原反应中,浓盐酸的量切不可过量,否则浓氨水用量将增加,最后导致溶液体积过大,造成产品损失。
[10]锡在还原作用中最终变成二氯化亚锡,它也溶于水。但加入浓氨水至碱性后,二氯化亚锡变成氢氧化亚锡沉淀可被滤去,而对氨基苯甲酸的盐酸盐成铵盐仍溶于水。
[11]如果溶液体积过大,则需要浓缩。浓缩时,氨基可能发生氧化而导入有色杂质。
[12]对氨基苯甲酸是两性物质,碱化或酸化时都要小心控制酸、碱用量。特别是在滴加冰醋酸时,须小心慢慢滴加。避免过量或形成内盐。
[13]为使产品少受损失,可采用分步抽滤的方法。即在有产品析出后,先滤集之,再将滤液加酸,如此反复抽滤,至无沉淀析出为止。
[14]酯化反应中,仪器需干燥。
[15]浓硫酸的用量较多,一是催化剂,二是脱水剂。加浓硫酸时要慢慢滴加并不断振荡,使之在反应液中分散均匀,以免加热引起碳化。
[16]酯化反应结束时,反应液要趁热倒出,冷却后可能有苯佐卡因硫酸盐析出。
[17]碳酸钠的用量要适宜,太少产品不析出,太多则可能使酯水解。因此,加碳酸钠粉末时要少量多次,每次加入后必须等反应完全后再可补加,切忌过量。
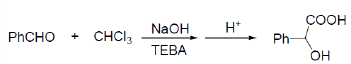
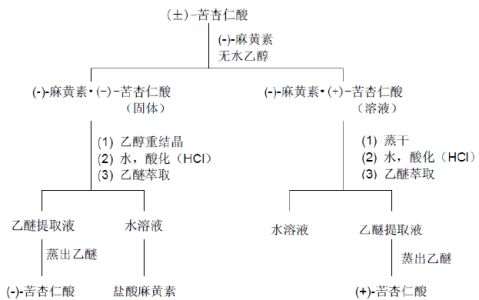
实验二:外消旋苦杏仁酸的合成与拆分(预备实验)
[反应式
]
[拆分流程
]
[试剂]
苯甲醛(新蒸),氯仿,三乙基苄基氯化铵(TEBA,自制),氢氧化钠,乙醚,浓硫酸,甲苯,无水硫酸钠,无水乙醇,盐酸麻黄素,浓盐酸
[步骤]
1. 三乙基苄基氯化铵(TEBA)的制备
在50 m1 装有回流冷凝管的圆底瓶中放入搅拌子,加入19 m1 1,2-二氯乙烷,在搅拌下依次加入5.5 m1苄氯、7 m1三乙胺,加热回流搅拌1.5 h。将反应物冷却,析出晶体,如不析出晶体,可用玻棒擦瓶壁促使结晶析出,抽滤,并用少量二氯甲烷或无水乙醚洗涤,烘干,产量约10 g。季铵盐易吸潮,干燥后的产品应置于干燥器中保存,或保存于塞紧的小锥瓶或试管中。
2. 外消旋苦杏仁酸的合成
在锥形瓶中小心配制13 g氢氧化钠溶于13 m1水的溶液,在水浴中冷却至室温。
在100 m1装有搅拌器[1]、回流冷凝管和温度计的三口瓶中,加入6.8 m1苯甲醛、0.7 g TEBA和12 m1 氯仿。开动搅拌,在水浴上加热,待温度上升至50~60℃,自冷凝管上口慢慢滴加配制的50%的氢氧化钠溶液[2],滴加过程中控制反应温度在60~65℃,约需45~60 min加完。加完后,保持此温度继续搅拌1 h[3]。将反应液用140 m1水稀释,每次用15 m1乙醚萃取两次,合并醚萃取液,倒入指定
容器待回收乙醚。此时水层为亮黄色透明状,用50%硫酸酸化至PH为1~2后,再每次用30 m1乙醚萃取两次,合并酸化后的醚萃取液,用无水硫酸钠干燥。在水浴上蒸去乙醚,并用水泵减压抽净残留的乙醚(产物在乙醚中溶解度较大),得粗产品6~7 g。将粗产物用甲苯进行重结晶(每克粗产物约需1.5 m1),趁热过滤,母液在室温下放置使结晶慢慢析出。冷却后抽滤,并用少量石油醚(30-60℃)洗涤促使其快干。产量:4~5 g。纯的外消旋苦杏仁酸为白色结晶,熔点118~119℃。
3. 外消旋苦杏仁酸的拆分
在50 m1锥形瓶中,将4 g麻黄素盐酸盐溶于10 m1 水,加入1 g氢氧化钠溶于5 m1水的溶液,摇荡混合后,(-)-麻黄素即游离出来。冷却后每次用10 m1乙醚萃取两次,合并醚萃取液并用无水硫酸钠干燥。在100 m1圆底烧瓶中蒸去乙醚后[4],即得(-)-麻黄素。
将上面制得的(-)-麻黄素溶于30 m1 无水乙醇,然后加入3 g苦杏仁酸溶于10 m1无水乙醇溶液,混合均匀后在水浴上隔绝潮气回流1.5~2 h。放置至室温让其自然结晶,然后在冰浴中冷却让其结晶完全。抽滤,粗产物用40 m1无水乙醇重结晶后得无色结晶约2.2 g,熔点165℃。重新用20 m1无水乙醇再结晶一次后,得到(-)-苦杏仁酸·(-)-麻黄素盐。产量:约1.5 g。纯的(-)-苦杏仁酸·(-)-麻黄素盐为白色粒状晶体,熔点169~170℃。
将上述得到的盐溶于10 m1水,然后用浓盐酸小心酸化至刚果红试纸变蓝(约需1 m1)。酸化后的水溶液每次用10 m1乙醚萃取两次,合并醚萃取液,经无水硫酸钠干燥后在水浴上蒸去乙醚,得(-)-苦杏仁酸白色结晶。产量:约0.5 g。纯的(-)-苦杏仁酸为白色结晶,熔点131~132℃。萃取后的水溶液倒人指定的容器内,以便回收麻黄素[5]。
将两次结晶(-)-苦杏仁酸·(-)-麻黄素盐后剩下的乙醇母液在水浴上蒸去乙醇,并用水泵将溶液抽干。残留物中加入20 m1水,温热并搅拌使固体溶解,然后小心用浓
盐酸酸化至刚果红试剂变蓝。此时若有油状粘稠物出现,可用滤纸滤掉。每次用10 m1乙醚萃取两次,合并醚萃取液,经无水硫酸钠干燥后蒸去乙醚,即得(+)-苦杏仁酸[6]。萃取后的水溶液倒入指定容器回收麻黄素。
4. 比旋光度的测定
将上面制得的(-)-苦杏仁酸准确称量后,用蒸馏水配成2%的溶液[7]。测定比旋光度,并计算拆分后单个对映体的光学纯度[2]。纯粹苦杏仁酸的[α]=±156℃。
[注释]
[1]也可用电磁搅拌代替电动搅拌,效果更好。相转移反应是非均相反应,搅拌必须是有效而安全的。这是实验成功的关键。
[2]浓碱溶液呈粘稠状,腐蚀性极强,应小心操作。盛碱的玻璃仪器用后要立即洗干净。
[3]此时可取反应液用试纸测其PH值,应接近中性,否则可适当延长反应时间。
[4]蒸出的乙醚可用于下一步萃取。
[5]将萃取后的水溶液在蒸馏瓶中蒸去大部分水,然后移至烧杯中浓缩至一定体积后,冷却结晶,抽滤析出的晶体,干燥,即可回收(-)-麻黄素。
[6] (+)-苦杏仁酸的分离显得更加困难,一般难以得到纯品。故建议安排学生实验时只分离对映异构体之一,即(-)-苦杏仁酸。
[7]如溶液混浊,需用定量滤纸过滤。
实验名称
重庆医科大学药物化学实验报告
日期: 院系: 药学院
时间: 专业:
天气: 年级: 学号: 学号: 学号: 评阅日期:
室温: 班别: 签字: 签字: 签字: 成绩:
湿度: 大组: 日期: 日期: 日期:
小组成员一, 姓名: 小组成员二, 姓名: 小组成员三, 姓名:
评阅教师签字:
左
侧装订区域内不得书写报告内容
纸张不够可以另外添加附页
药物化学实验记录首页
左侧装订区域内不得书写报告内容 纸张不够可以另外添加附页 实验名称
续页—药物化学实验 第 页