(1)吸入增加CO2的氣體→呼吸運動加深加快。
呼吸頻率加快是由於吸入空氣中PCO2增加,使得血液中PCO2增加,CO2通過血腦屏障進入腦脊液中溶於水,在碳酸酐酶的作用下分解成HCO3- + H+ , H+刺激延髓化學感受器,間接作用於呼吸中樞,通過呼吸肌的作用使呼吸加強。PCO2增加還刺激主動脈體和頸動脈體外周化學感受器,反射性的使呼吸加深加快。
(2)缺氧→呼吸運動加強。
吸入氮氣造成肺泡氣中氧分壓降低,而由於CO2擴散快,故肺泡PCO2基本不變,血液中氧分壓下降,使外周化學感受器興奮;低氧對呼吸中樞的直接作用是抑制性作用,但輕、中度缺氧時,興奮作用大於抑制作用使呼吸中樞興奮,呼吸運動加強。重度缺氧時抑制作用為主,出現呼吸抑制。
(3)增大無效腔→增加氣道長度後家兔呼吸張力增加,呼吸頻率增加。
增加氣道長度等於增加無效腔,增加無效腔使肺泡氣體更新率下降,引起血中PCO2、PO2-下降,刺激中樞和外周化學感受器引起呼吸運動會加深加快;另外,氣道加長使呼吸氣道阻力增大,減少了肺泡通氣量,反射性呼吸加深加快。
(4)乳酸酸中毒(血液中H+增高)→呼吸運動加深加快。
靜脈注射乳酸後,改變了血液中的PH值,血液[H+]↑,H+是化學感受器的有效刺激物,它可以通過刺激外周化學感受器調節呼吸運動,也可以通過血腦屏障後刺激中樞化學感受器而起作用。但因為H+不易通過血腦屏障,故血中H+對中樞化學感受器直接刺激作用不大,主要還是刺激外周感受器。
(5)靜脈注射嗎啡→呼吸運動減慢。
嗎啡能抑制大腦呼吸中樞的活動,降低其對CO2張力的敏感性,並可抑制呼吸調整中樞,使呼吸頻率減慢。急性中毒會導致呼吸中樞麻痹、呼吸停止至死亡。
(6)靜脈注射尼可刹→呼吸加強。
尼可刹米主要是興奮延腦呼吸中樞,也可刺激頸動脈體和主動脈體化學感受器,反射性興奮呼吸中樞,並能提高呼吸中樞對CO2的敏感性。
(7)剪斷迷走神經→切斷一側迷走神經後,動物的呼吸運動呈快而淺。 切斷雙側頸迷走神經後,動物的呼吸運動呈慢而深。 由於一側迷走神經的神經衝動傳遞受阻,使得呼吸運動的調節受阻;而迷走神經為混合神經,另一側迷走神經將起到呼吸調節作用,發揮負反饋調節,加速吸氣和呼氣活動的交替。
當切斷兩側迷走神經後,中斷了肺牽張反射的傳人通路,肺牽張反射的生理作用被消除,因而呈現出慢而深的呼吸運動,使吸氣延長。【迷走神經中含有肺牽張反射的傳人纖維。肺牽張反射中的肺擴張反射在於阻止吸氣過長過深,促使吸氣及時轉人呼氣,從而加速了吸氣和呼氣動作的交替,調節呼吸的頻率和深度。】 實驗結論
1. 機體通過呼吸調節血液中的O2、CO2、H+水準,動脈血中O2、CO2、H+的變化又通過化學感受器調節呼吸,維持機體內環境的相對穩定。迷走神經是呼吸運動調節反射中的傳入神經,剪斷一側迷走神經後,可通過反饋調節使呼吸變淺,頻率加快。
2. 注射嗎啡要緩慢,邊注射邊觀察,當出現呼吸明顯抑制時,立即停止注射
3. 耳緣靜脈穿刺時,因觸動兔耳及針刺激可能引起家兔呼吸改變,故需待這些變化消失,呼吸恢復正常後,再向靜脈內推注藥液
第二篇:第五章 影响药物吸收的生理因素
第五章 影响药物吸收的生理因素
体内药物的吸收取决以下三方面:药物的理化性质;药物的特性;药物的吸收部位的构造和生理功能。它们在制剂生产和生物药剂学评价中非常重要(第六章)。本章重点讲述的是药物在体内药物吸收的生理因素的影响。药物在吸收过程中的生理和病理因素,对药物的选择以及避免可能的药物与药物、药物与食物之间的相互作用是非常重要的。
1.细胞膜的性质
在药物吸收过程中,药物分子必须从吸收部位穿过一层或更多的细胞膜进入体循环。药物从吸收部位进入体循环的透过能力与药物的分子结构和细胞膜的物理和生化性质有关。药物必须透过细胞膜才能从吸收部位进入细胞。
胞内转运吸收是药物穿过细胞膜的一种方式。一些极性分子不能穿过细胞膜,但是能透过细胞间隙,这种透过方式被称为胞间转运吸收。图5-1描述了这两种方式的不同之处。有些药物可能是通过一种或多种的混合方式被吸收的。
细胞膜是细胞的一种重要结构,它把包围整个细胞(血浆原生质膜),并且起到细胞和组织间液的边界作用。此外,细胞膜包裹绝大多数的细胞内器官(例如,线粒体)。从功能上来说,细胞膜是对分子的透过是一种选择性的屏障。细胞膜是半透膜,水、一些小分子和脂溶性分子能透过,但如蛋白质和结合了蛋白质的药物这些带大量电荷的分子和大分子量的物质是不能够透过。
图5-1 小肠上皮细胞转运载体。长方形和椭圆形分别代表主动转运和促进转运载体。主动转运载体的同向箭头表示物质的同向转运力。胞内转运吸收;胞间转运;小肠上皮细胞;刷状膜侧;基膜侧;钠/氨基酸;氨基酸;钠离子,氯离子/β氨基酸;氢离子/寡肽;钠离子/D葡萄糖;D果糖;氢离子/乳酸;碳酸根离子;一元羧酸;钠离子/磷酸;钠离子/胆汁;氢离子/硝酸;碳酸氢根/硝酸;氢氧根离子/叶酸;胆碱;糖蛋白;钠泵;己糖;钾泵。
药物的跨膜转运受到细胞膜的组成与结构的影响。细胞膜一般是很薄,大约70~100埃。细胞膜主要由磷脂、散布的糖和双层蛋白质所组成。人们提出了膜结构的一些假说。1952年Davson与Danielli最开始提出了脂质双分子层理论,认为细胞膜是磷脂质双分子层结构构成,蛋白质分布在其两侧,脂质分子的亲水基分布在膜的外侧,疏水基排列在膜内部。脂质双分子层假说解释了为什么脂溶性药物分子比极性药物分子更容易透过细胞膜,但并不能解释为什么水、小分子如尿素和带电离子可以透过细胞膜。
1972年Singer和Nicolson提出了液态镶嵌模型,解释了极性分子的胞间转运。根据此模型,双层脂质膜为基质,球形蛋白镶嵌其中,这些球形蛋白是一些极性分子和带电离子的透过脂质屏障的选择性通道。如图5-2所示,跨膜转运蛋白交互分布在整个生物膜上,根据对毛细管膜转运的研究(1990,Pratt与Taylor),得出生物膜上有10nm和50~70nm的两种小孔,水分子、离子、溶质分子如尿素等从这些小孔透过细胞膜。
图5-2,质膜的模型组成包括蛋白、糖类和脂类。膜内在蛋白质嵌入在质膜中,膜周边蛋白质仅仅结合在膜表面。糖类由单糖和多糖以链状结合而成,它与蛋白结合称之为糖蛋白,与脂质结合称之为糖脂。膜的不对称存在以下几个方面。糖类多分布于外表面,膜周边蛋白质多分布于细胞质侧或内表面。双层的脂质膜由具有不同特征的不许多种类的脂质分子组成。更为重要的是,每一类膜内在蛋白质具有确定的转运方向,它所对应的分子同样如此。
2. 药物的跨膜转运
2.1被动扩散
被动扩散指的是物质从膜的高浓度向低浓度一侧的自发转运过程,此过程因为不需外部能量所以是被动的。图5-3描述了药物分子往返透过生物膜。当膜两侧药物分子浓度相等,往返的药物分子会达到平衡,则药物的净转运为零。当膜一侧药物浓度较高,在任何给定时间,该侧药物分子进入的就比出来的多,结果药物分子从高浓度一侧转运到了另一侧,如图大箭头所示。这种转运的速度被称为流速,由矢量表示。分子自然的会向各个方向运动,因为由分子具有动能并且在空间彼此碰撞。图5-3仅描述了分子向左和向右的运动,因为向其它方向的运动分子受器壁的限制而不能影响浓度改变。
大多数药物是通过被动扩散被吸收的。被动扩散的驱动力来自于药物在黏膜侧相对于在血液中的高浓度。根据Fick‘s扩散定律,药物分子从高浓度一侧扩散至低浓度一侧的速度表示如下:
dQ/dt= DAK/h (CGI-CP) (5.1)
式中:dQ/dt为扩散速度;D为扩散常数;K 为油水分配系数;A为膜表面积;h为膜厚度;CGI-CP是药物在胃肠道和血浆中的浓度差
因为药物在进入血液后,很快分布在较大容积中,与吸收部位相比血液中的药物浓度很低。例如,药物常给予数毫克的剂量,然而血浆中的药物浓度经常在µg/ml或ng/ml的数量级范围内。如果是口服给药,CGI>>CP,存在一个较高的浓度梯度,使药物从胃肠道中扩散至血浆中。
根据Fick‘s扩散定律,影响药物被动扩散的还有另外一些因素。例如,药物的脂溶性大小可会影响药物的吸收。油水分配系数K代表的是药物在油相和水相间的分配,脂溶性大的药物其K值也大。膜的表面积也影响吸收的速度,药物可以从胃肠道的大部分表面被吸收。但在小肠的十二指肠部位存在药物的最大吸收,这是因为具有很大表面积的绒毛和微绒毛,而这些绒毛在胃肠道的其它部位较少。
在一定的吸收部位,假想膜的厚度h是常数。与药在脑中毛细血管细胞膜的扩散相比,药物在其他部位毛细血管细胞膜的扩散较快。脑部毛细血管和神经胶质紧密结合,像有一层厚的脂膜,使得药物缓慢扩散进脑中。血脑屏障指的就是水溶性分子通过毛细血管细胞膜进入脑的较差的扩散性。但在某些疾病状态下,这些细胞膜可能被破坏,而使药物容易透过。
扩散系数D对一种药物来说是一个常数,是当浓度梯度一定时单位时间通过单位面积膜的药物的量。D的量纲是面积每时间单位,例如cm2/sec。
因为一定的吸收过程中D,A,K和h都是常数,组合常数或渗透系数P可以定义为:
P=DAK/h (5.2)
在公式5.1中,药物在血浆中的浓度CP与在胃肠道中的浓度CGI相比非常小。若忽略CP,并且将P带入到公式5.1中,便能得到如下关系式:
dQ/dt= P(CGI) (5.3)
公式5.3是一级吸收速率表达式。实际上,大部分药物的血管外吸收都是一级吸收过程,而且由于CP 和CGI之间较大的浓度差,药物吸收速度一般要比消除速度快。
许多药物既有亲水基团,又有亲油的基团。脂溶性大的药物比脂溶性小或水溶性大的药物更易透过细胞膜。对于弱电解质的药物,如弱酸、弱碱,离子化程度影响药物转运。离子化药物带有电荷更溶于水,而非离子化药物脂溶性更大。弱电解质的离子化程度取决于药物的pKa和溶液的pH。Henderson和Hasselbalch用如下表达式描述了弱酸或弱碱的离子化程度与pKa、pH之间的关系。
对于弱酸 盐/酸=(A-)/(HA)=10(pH-pKa) (5.4)
对于弱碱 碱/盐=(RNH2)/(RNH3+)=10(pH-pKa) (5.5)
根据公式5.4和5.5,若已知药物的pKa,在给定pH的情况下,可以求出游离型与解离型药物的比例。例如,血浆pH为7.4,水杨酸(pKa3.0)大部分以解离型的形式存在,如下所示。
 盐/酸=10(7.4-3.0)
盐/酸=10(7.4-3.0)  log(盐)/(酸)=7.4-3.0=4.4 盐/酸=2.51×104
log(盐)/(酸)=7.4-3.0=4.4 盐/酸=2.51×104
假设药物分配只受Fick‘s扩散定律的影响,那么药物在膜两侧达到平衡时的浓度应相等。对于非电解质或非解离型的药物,平衡时在膜两侧的浓度相等,但对于电解质或可电离的药物,当膜两侧的介质pH不同时,在平衡时膜两侧的药物浓度不同。例如,水杨酸(pka3.4)在胃液中(pH1.2)的浓度与在血浆中(pH7.4)的浓度不同。(图5-4)
图5-4,口服弱电解质药物的分配模型
根据Henderson-Hasselbalch公式,水杨酸在pH7.4和pH1.2下将以如下比例存在:
在血浆中(pH7.4) (RCOO-)/(RCOOH)=2.51×104
在胃液中(pH1.2) (RCOO-)/(RCOOH)=10(1.2-3.0)=1.58×10-2
在不同pH下,水杨酸在膜两侧总浓度见表5.1。因此pH影响水杨酸(RCOOH)和水杨酸盐(RCOO-)的透膜分布。假定RCOOH可以自由透过细胞膜,而RCOO-不能透过细胞膜,在这种情况下,水杨酸分配平衡时在血浆中的浓度比在胃液中的浓度高出约25000倍(表5.1)。根据公式5.5,同样可以计算的弱碱的分配比率。
表5.1 水杨酸在不同pH下的相对浓度
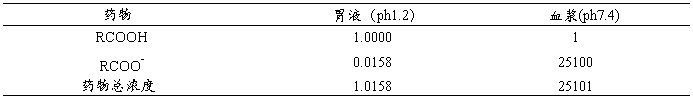
根据pH分配假说,假如细胞膜两侧的pH不同,那么(1)弱酸或弱碱药物在各自膜的一侧发生不同程度的解离;(2)膜两侧总的药物浓度(包括电离和未电离的)不相等;(3)解离程度高一侧总的的药物浓度高。因此弱酸性药物如水杨酸在胃中(pH1.2)吸收很快,而弱碱性药物如奎尼丁在胃中吸收很慢。
影响药物在膜两侧浓度的另一个因素是药物对组织的特殊亲和力。这种亲和力能够阻碍药物在膜两侧的自由扩散。例如,药物可能与血浆或组织蛋白结合,这些药物-蛋白结合体多见于双香豆素类、磺胺类药物等。 而且,像氯丹这种脂溶性杀虫剂可溶解在脂肪组织中。另外,四环素类的药物可与骨骼与牙齿中的钙形成复合物。最终由于主动转运过程或特定的吸收过程,使药物在组织中蓄积。这些过程已经被甲状腺中的碘化物、细胞浆中的钾以及肾上腺素存储位点的儿茶酚胺的蓄积所证明。
2.2载体转运
理论上脂溶性药物能通过细胞或分布在细胞周围。对小分子量的脂溶性药物,脂质细胞膜不会成为药物扩散和吸收的屏障。分子量小于500MW的药物可以经胞间转运被吸收。人们研究了体内大量的载体转运系统,尤其是胃肠道的离子和人体所需营养物质的吸收中的载体转运系统。
2.2.1主动转运
主动转运是载体介导的跨膜转运,它在药物在胃肠道的吸收、药物及其代谢产物在肾脏和胆汁的排泄过程中起着重要作用。少数和生理代谢产物相似的脂溶性药物以主动转运的方式在胃肠道中被吸收。主动转运的一个特点是逆浓度梯度转运,即从低浓度一侧向高浓度一侧转运,因此,此过程消耗能量。另外,主动转运需要载体,载体与药物形成复合物并穿过细胞膜,在膜的另一侧解离释放药物(图5-5)。
图5-5 载体转运示意过程 胃肠道 小肠上皮细胞 血液 药物 载体 复合物
载体对药物分子具有高选择性。与发生主动转运的底物在结构上相似药物,很可能以相同的载体转运机制被转运。因此结构相似的药物会互相竞争载体上的结合位点,又由于可利用的载体数量是一定的,如果药物的浓度很高,载体上的结合位点就被饱和。图5.6比较了药物吸收速度和药物在吸收部位浓度的关系。由图注意到,被动扩散的药物吸收速度与药物浓度呈线性关系。相反,载体转运的药物,药物吸收速度随其浓度的增加而增加直到载体分子被完全饱和。当药物浓度过高时,药物吸收速度保持为一常数不便。
2.2.2. 促进扩散
促进扩散也是一种载体转运方式,与主动转运的不同之处是药物沿浓度梯度的方向转运(由高浓度一侧向低浓度一侧),因此这种转运方式不消耗能量。但是,由于这种方式是由载体介导的,它也可以被饱和,也具有对药物的结构选择性,结构类似的药物表现出竞争动力学特征。在药物的吸收过程中,促进扩散不是主要的。
2.2.3. 载体介导的肠道转运
在小肠绒毛的刷状缘、底膜和侧膜部位,存在许多载体转运系统,用来吸收特殊离子和人体重要的营养物质(Tsuji和Tamal,1996年)。许多药物因为和体内的底物结构类似,能被底物相应的载体转运(表5.2)。人们在小肠内已发现一种跨膜转运蛋白─P-糖蛋白(P-gp),P-糖蛋白能明显减弱多种亲油性和细胞毒性药物透过小肠上皮细胞的能力。小肠内还发现其他的转运载体(Tsuji和Tamal,1996年)。例如,许多口服的头孢菌素通过氨基酸转运载体被吸收。头孢唑林是一种非胃肠道给药的头孢菌素类药物,因为通过这种机制吸收程度很少故口服无效。
图5-6 被动扩散(A)和载体转运(B)的吸收速率比较。
表5.2 小肠转运载体以及转运的药物

2.3囊泡转运
囊泡转运是细胞吞噬微粒或溶解于其中的物质的一种转运过程。根据摄取物质的种类,囊泡转运分为胞饮作用和吞噬作用两种。胞饮作用是指细胞吞噬溶质或溶液分子,而吞噬作用是指利用巨噬细胞吞噬大的微粒和大分子。出胞和入胞分别是大分子进出细胞的两种过程。
在胞饮或吞噬过程中,细胞膜在微粒处不断下陷,最后把其吞入细胞内(图5-7)。吞饮了微粒的细胞随后在其内部形成囊泡。囊泡转运被认为是口服给药的疫苗和多种生物蛋白类药物的吸收过程。
图5-7 出胞入胞示意图 出芽
蛋白质的转运是胞吐作用的一个例子,如胰岛素从胰的胰岛细胞中产生转运到胞外。首先胰岛素被包裹进入囊泡内,然后与细胞膜融合,释放出胰岛素。
2.4膜孔转运
很小的分子如尿素、水和各种糖等能很快透过细胞膜,就像膜上有通道或膜孔。尽管在显微镜下并没有观察到这些膜孔,但是药物分子通过水性孔道的模型常用来解释药物的肝吸收和肾排泄过程。
一种被称为转运蛋白的蛋白质可形成了脂膜上的开放通道。包括药物在内的各种小分子以扩散的方式通过通道时,比在膜上其他位置通过时更快。
2.5离子对的形成
强电解质药物被高度离子化而带有电荷,例如pKa值很大的四价氮化合物。在各种生理pH下,强电解质药物带有电荷,透膜性极差。当离子化的药物与带有相反电荷的离子结合后,就形成了电中性的离子对。这种电中性的复合物容易通过细胞膜。例如,普奈洛尔与油酸形成离子对后被吸收,有利于普奈洛尔的吸收,还有奎宁与已基水杨酸盐可以形成离子对。
3.给药途径
经非胃肠外、小肠、吸入、透皮、鼻黏膜途径给药有各自的优缺点。表5.3列出了常用给药途径的特点。许多药物因为它们在胃肠道中不稳定或被小肠中的消化酶降解,不能口服给药。例如红细胞生长素和人生长激素都是以肌肉注射给药。胰岛素由于在肠道中被降解,以皮下给药或肌肉注射的方式给药。药物经皮下注射的吸收速度药慢于经静脉注射的吸收速度。经非胃途径给药的生物利用度和药效受到给药部位血流量和疾病因素的影响。制剂的生物利用度将在第十章讨论。生物制品在胃肠道内太不稳定而不能口服给药。
表5.3 常用给药途径特点

实用焦点
1994年,Jensen JD研究了尿毒症患者和健康受试者的红细胞生长素(EPO)的药动学特点。在皮下注射给药后,病人的生物利用度明显低于健康受试者(23.7%:38.5%;p<0.01),而且患者血药浓度很低(113:153U/L;P<0.05),达峰时间比健康受试者延迟(15.4:110h;p<0.02)。
皮下注射是生物利用度低的药物除口服给药途径外的另一种给药方式。例如,西马地平在口服给药时由于广泛的代谢作用和较小的吸收,故生物利用度只有14%,而皮下注射西马地平可以很快吸收,可;立刻缓解偏头痛。
3.1口服药物吸收
3.1.1. 解剖和生理因素
消化系统由从口腔到肛门的消化管组成在。在胃肠道(GI)中发生的主要生理过程包括分泌、消化和吸收。分泌是消化液(包括溶液、电解质、多肽和蛋白质等)进入消化管的过程。糖和蛋白质的消化过程与唾液中的酶和胰液有关。其它的分泌液,如黏液,能保护胃肠道管内壁。消化是将食物变成小的团块从而有利于吸收。食物主要在小肠的近端(十二指肠)被吸收,吸收是消化道中的成分进入体内血液以循环中。吸收可以看作是物质在消化道和血管之间转运的净结果。
药物口服后经过消化道的各个部位,包括口腔、食管和胃肠道。物质残渣最终通过肛门排到体外。总的转运时间包括胃排空、小肠转运和结肠转运等的时间,约在0.4~5天(Kirwan 和Smith,1974年)。小肠是药物吸收最重要的部位。对于健康者来说,小肠转运时间(SITT)为3~4小时。如果药物在离开小肠时还未被完全吸收,那么它的吸收较差。正常情况下,小肠里充满了消化液,使其内容物保持流动性。相反,在结肠中消化液被重吸收,其内容物是固体或半固体状态,使得药物的溶解变差或较困难。具有溶解作用的食糜和消化液的缺乏会造成对药物吸收不利的环境。
消化道的正常生理功能可能后受到食物、胃肠道内容物、激素、内脏神经系统、疾病和药物影响,因此口服药物的吸收可能受到生理结构、功能和消化管中内容物的影响。而且,药物本身的理化性质和药理特点也影响其自身的吸收。
(1)口腔 唾液是口腔内主要的分泌液,pH值约为7,含有能降解淀粉的唾液淀粉酶。粘蛋白是口腔分泌的一种糖蛋白,可湿润食物,能与药物发生作用。人每天唾液分泌量大约是1500ml。
(2)食管 食管连接咽和胃的幽门部。食管中液体的pH值在5~6之间。食管的下端是食管括约肌,能防止胃酸倒流。片剂或胶囊服用后可能停留于此引起刺激。只有少量的药物在食管里溶出。
(3)胃 胃受迷走神经支配。但除此之外,局部神经网、激素、胃肠道壁机械性刺激感受器的敏感性和化学受体等还可以调节胃的分泌,包括对胃酸和胃排空的调节。禁食状态下,胃中的pH值在2~6之间。在胃中有食物的情况下,由于壁细胞分泌胃酸,pH降至1.5~2.0。胃泌素和组胺能刺激的胃酸分泌,胃泌素由G细胞分泌,它主要存在于胃黏膜和十二指肠中。胃泌素的释放受胃的舒张以及肽和氨基酸的调节。一种与VB12的吸收有关的内在因子和各种胃酶被分泌进入胃中产生消化过程,如胃蛋白酶可消化蛋白质。
碱性药物在胃酸中溶解很快。胃研磨指将大的食物颗粒充分混合,其受到胃室的挤压又被分散成小颗粒。食物和液体通过幽门括约肌的扩张进入十二指肠。食物的种类和粘度影响胃排空。脂肪酸和单、双甘油酯延缓胃排空(Hunt和Knox,1968粘)。粘稠的食物一般胃排空较慢。胃排空的时间和药物吸收之间的关系将在下章展开讨论。
(4)十二指肠 胰和胆囊通过一条的导管与十二指肠相连。十二指肠中有碳酸氢盐中和从胃排出的酸性食糜,它的pH值约为6~6.5。此pH对消化蛋白质和肽类食物的蛋白酶很有利。含酶胰液分泌后通过胆管进入十二指肠。胰蛋白酶、胰凝乳蛋白酶都参与蛋白质被水解成氨基酸。淀粉酶参与糖类的消化。胰脂肪酶分泌液将脂肪水解成脂肪酸。十二指肠中复杂的液体介质有助于水溶性差的药物。
十二指肠是许多酯类前药被水解吸收的部位。蛋白质酶使得许多蛋白质类药物在十二指肠中不稳定,阻碍蛋白类药物的充分吸收。
(5) 空肠 空肠是小肠的中间段,介于十二指肠和回肠之间。利用从十二指肠进入的胰液和胆汁,蛋白质和糖类进一步被消化。一般空肠的收缩性比十二指肠弱,常被用来研究药物的体内吸收。
(6)回肠 回肠是小肠的终端。回肠的收缩性比十二指肠弱,常被一些膨胀球体所堵塞,回肠被灌注后可用于研究药物的吸收。回肠里的pH约是7,在回肠末端可达8。由于回肠分泌碳酸氢盐,酸性药物可被将溶解。胆汁有助于溶解脂肪和亲油性药物。回肠与盲肠的瓣膜将小肠和结肠离开。
(7)结肠 结肠缺乏绒毛,且由于其内容物为粘度大的和半固体物质,使得药物在结肠的吸收差。结肠内也有和黏液素一样的物质,具有润滑和保护肠壁的功能。结肠内的pH范围是5.5~7。只有很少的药物在回肠里被吸收,如茶碱。能在回肠里被充分吸收的药物可以制成口服缓释制剂。结肠里存在代谢某些药物的厌氧菌和需氧菌。例如左旋多巴胺和半乳糖苷果糖能被结肠菌群所代谢。结肠疾病能够影响结肠,使其肠壁增厚,使微生物系变得更加厌氧。在这种疾病下,氯林可霉素和普奈洛儿的吸收增加,但其他的药物吸收减少(Rubinstein等,1988)。
(8)直肠 直肠长约15cm,终止于肛门。如果没有粪便,直肠里只有约2ml pH约为7的液体。直肠分布有上直肠静脉、中直肠静脉和下直肠静脉,下直肠静脉(靠近肛门括约肌)和中直肠静脉下腔静脉的血液不经肝返回心脏。上直肠静脉通过门静脉,进入肝脏。
直肠给药后,药物的吸收量根据栓剂位置或药物的溶解度不同而不同。一部分药物经过下直肠静脉直接吸收进入体循环。一些药物通过上直肠静脉进入门静脉到达肝脏,经代谢后被机体吸收。
3.2药物在胃肠道中的吸收
药物可通过各种途径以被动扩散被吸收,包括舌下、口腔、胃肠道和直肠。对大多数药物,药物口服后吸收的最适宜部位是在小肠的上部或十二指肠。十二指肠独特的结构为药物的被动扩散提供了一个较大的表面积(图5.9)。十二指肠巨大的表面积是由于在粘膜上有褶皱,而褶皱存在被成为绒毛的小突起。这些绒毛还有的微绒毛,形成了一种刷状膜。另外,十二指肠上布满了许多毛细血管网。这些毛细血管能够保持药物在肠道和血液循环的浓度梯度。
图5.9 三种增加小肠吸收表面积的机制:褶皱、绒毛,微绒毛。
3.2.1胃肠道的蠕动
药物口服以后,在胃肠道中的确切位置及环境是很确定。胃肠道的蠕动使药物通过消化道,而不是总停留在吸收部位。对口服药物来说,在胃肠道中存在能有效吸收药物的吸收窗。药物可以从非生物降解的控释制剂中完全释放到此吸收窗中被吸收,这种吸收方式要比制剂在整个肠道中吸收更好。
药物在胃肠道中的转运时间取决于药物的药理学性质、药物的剂型和各种生理学因素。药物在胃肠道中的生理运动取决于消化道中是否有刚吃下的食物(消化或进食状态)或是处于禁食状态还是正消化状态(图 5-10)。在禁食或消化状态下,交替循环运动起到了推动作用,使胃肠道上部到盲肠能够排空。最初,消化道是静止的,然后是不规则收,接下来较大幅度有规律收缩,推动着那些残留内容物到消化道的远端。在进食状态下,无规则的收缩代替了交替循环运动,这种无规则收缩起到混合肠道内容物的作用,并且加快了肠内容物分为许多小部分向结肠方向的流动(表 5.4),幽门和回盲的瓣膜防止食物从远端到近端的回流。
表5.4 禁食状态下狗体内的蠕动方式特点

3.2.2 胃排空时间
从解剖学上看,吞咽的药物能迅速到达胃部,然后通过胃排空到达小肠。由于十二指肠吸收能力较强,延缓胃排空时间,从而延迟药物到达十二指肠,这样可能会降低药物的吸收速度和程度。因此,延缓了药物的起效时间。一些药物,如青霉素,在酸性环境中是不稳定的,如果胃排空时间延长就会降解;另一些药物,如阿司匹林,延长胃接触时间会引起胃粘膜刺激。
有许多因素可以影响胃排空时间(表 5.5)。延缓胃排空的因素包括进食高脂肪的食物、冷饮料和抗副交感神经药物(Burks等 1985年;Rubinstein等 1988年)。液体和小于1毫米的颗粒通常不能在胃中滞留。通常以为这些小颗粒由于在胃具有比十二指肠稍高的基底压力而被排空。进食不同成分的食物使胃排空速率也不同。Feldman及其合作者(1984年)观察到10盎司软饮料,搅匀的鸡蛋(可消化的固体)和辐射透不过的物质(不可消化的固体)在胃中排空50%的时间分别是30分钟、154分钟和3-4小时。因此,在胃中液体通常比可消化固体排空快(图 5-11)。大颗粒,包括片剂和胶囊,在食物的存在的条件下胃排空时间为3-6小时。在消化间歇期,没有食物的存在,胃蠕动性较差,依靠节律性收缩排空内容物,不可消化的固体排空很慢(图 5-12)。
图5.11 同位素标记法研究胃排空。饭后在不同的时间间隔测定留在胃中的同位素百分数。从图中可看出液体排空呈指数关系,而固体排空呈线性关系。
3.2.3小肠蠕动
通常蠕动会混合十二指肠中的内容物,使药物颗粒与肠粘膜细胞紧密接触。药物必须在吸收部位有足够的时间停留以达到最大吸收。在肠道快速蠕动的情况下,像痢疾,药物停留时间很短,充分吸收机会较小。
在口服乳果糖(结肠菌群发酵产生的)后,早期研究发现,应用氢检测的的间接法,得出平均正常的小肠转运时间(SITT)大约是7个小时。近期研究用β射线扫描法的发现SITT大约为3-4小时。因此,在禁食状态下,药物通过胃和小肠时间是4-8小时。在进食状态下,SITT约为8-12小时。对于缓控释制剂,它们使药物延缓至很长时间内释放,此种剂型必须停留在肠道中的一定部位,使药物能在制剂被排泄出去之前释放并吸收。小肠转运将在第7章缓释制剂的设计中作进一步讨论。
3.2.4胃肠道的血液循环
胃肠道的血流对于转运已吸收的药物到体循环是很重要的。在十二指肠区和腹膜部,分布有广泛的毛细血管网和淋巴管。内脏血循环大约占心输出量28%,且在饭后还会增加。药物在小肠中被吸收后,经肠系膜管到肝门静脉和肝脏,然后才到达体循环。任何小肠血流量的减少如充血性心衰,将减弱药物在肠道中的转运,从而降低药物的生物利用度(Benet 等,1976年)。
淋巴循环在药物吸收中的作用也很重要的。药物通过细胞表面的微绒毛下的淋巴管而被吸收。药物通过淋巴吸收可不进入肝门静脉,从而避过由肝的首过效应。淋巴管对于油脂性食物的吸收很重要,一部分亲脂性药物可由此被吸收。许多水溶性差的药物可溶于油和脂中,这些药物可溶解在乳糜微粒中且经淋巴循环被吸收。把博来霉素载入乳糜微粒可以通过淋巴循环提高口服吸收。(Yoshikawa等,1983,1989)。
表5.5影响胃排空的因素
3.2.5食物对胃肠道药物吸收的影响
食物存在于胃肠道中可以影响药物的生物利用度。可消化的食物包含氨基酸、脂肪酸和许多营养物质,它们可以影响小肠的PH值和药物的溶解度。食物的影响并不确定。对于一些抗生素,例如青霉素、四环素,由于食物使吸收降低。而其他药物,如灰霉素,当进食一些高脂肪食物时,可以更好地被吸收(图 5-13)。胃肠道中食物的存在时可以刺激胆汁的分泌。胆汁中包含消化和溶解脂肪的表面活性剂-胆酸,并且可以通过形成胶团提高脂溶性药物的溶解度。对于一些碱性水溶性较差的药物,胃中的食物可以刺激盐酸的分泌,从而降低PH值,使药物更快的溶解及更好地吸收。当胃酸分泌减少时,碱性药物的吸收会降低(Ogata等,1986年)。
图5.13 进食不同类型的食物对服用灰黄霉素1.0g后血药浓度的影响比较。
通常对于处于禁食状态下或饮用了大量的水后的病人,药物的生物利用度较高(图 5-14)。但是一些药物如抗生素、铁盐、阿司匹林和非甾体类抗炎药(NSAID)对胃粘膜有刺激性,应和食物一起服用,这样会降低吸收的速度,但不改变吸收程度。
图5.14 健康禁食的受试者服用阿司匹林650mg(图A)、红霉素500mg(图B)、阿莫西林500mg(图C)、茶碱260mg(图D),送服水的多少的影响。
药物的一些剂型也会受食物的影响。食物延迟了胃排空,肠溶衣片将在胃中滞留较长时间。因此,肠溶衣片不能很快达到十二指肠,延迟了药物的释放和吸收。相反,肠溶微球或微丸分散在胃中,它们的排空很受食物影响较小,并且在十二指肠药物有更稳定的吸收。
食物也可以影响制剂的完整性,从而使药物的释放产生变化。例如,服用茶碱的24小时控释片,在进食状态下其生物利用度比禁食状态下更高(图 5-15)。
图5.15禁食和早餐后服用1500mg剂量的茶碱24小时控释片的药时曲线。阴影区表示病人恶心、呕吐或头痛的阶段。摄取食物后药物的释放与“药物储库”一致。
3.2.6 双峰现象
一些药物如雷尼替丁、西咪替丁、双嘧达莫口服后的药时曲线存在两个峰(图 5-16)。这种双峰现象常见于禁食病人服用单剂量后。双峰现象的理论基础是胃排空的变化、肠运动的变易、食物的存在、再循环及剂型的失误。
西米替叮双峰现象(Oberle和Amidon,1987年)可能是由于单剂量给药后在整个吸收过程中胃排空和肠蠕动速率的变化。对大多数药物来说,在胃部吸收很少。对于水溶性好的药物,在胃中溶出,部分被排空到十二指肠导致第一个吸收峰。随后药物排空到十二指肠,这种延迟产生了第二个吸收峰。
相反,某些药物在口服或非胃肠道给药后产生双峰现象,其给药后通过体循环明显地集中于胆囊的胆汁中。当受到食物刺激时,胆囊收缩并且将含药的胆汁释放到小肠中,药物被重吸收和再循环。
片剂的完整性可能也是产生双峰现象的一个因素。Mellinger 和Bohorfoush(1966年)在志愿者中对比了双嘧达莫完好和碎裂的药片,发现没有崩解和崩解不完全的片剂可以延缓胃排空从而产生第二个吸收峰。
图5.16 双嘧达莫的血清浓度(四人一组,共三组)。A 服用25mg 剂量的完整片剂;B 服用破碎的片剂;C 午饭后2小时服用完整片剂。
3.3实用焦点
3.3.1 食物对给药的影响
许多抗生素,例如青霉素、四环素,当饭时服用会使吸收延迟或降低,药剂师通常建议患者饭前1小时或饭后2小时以避免吸收的延迟。油性食物可以延长胃排空时间使其超过2小时,所以吃过油腻性食物的患者如果可能的话应在饭后3小时或更长的时间后用药。食物可使胃对肠溶衣片或不崩解制剂的排空延迟数小时,细小颗粒(小于1~2毫米)和在胃中崩解的片剂的胃排空不会受到食物的影响。
液体可以使胃扩张,加速胃排空,但是具有高热量的营养液延迟胃排空。吸收的降低有几种原因引起,,例如:牛奶和其他含钙的食物由于螯合作用使四环素吸收降低。由pH值的升高而使溶出减慢更能显著的减少吸收。1971年Barry 与Garrettson 报道,同时服用碳酸氢钠使胃pH值升高,减小了四环素的溶出合吸收。
饭后2小时,食物也能药物的吸收。例如:Borin等19995年对20个成年人进行头孢多星的吸收受油性食物的影响实验,结果与空腹时或饭前1小时给药相比,饭时和饭后2小时给药药时曲线下面积(AUC)与峰浓度(Cmax)均有显著提高。达峰时并不受食物的影响,表明食物增加了吸收的程度,但是没有增加吸收的速度。结果显示饭时或饭后服用头孢多星课使其吸收提高。
噻氯匹定是一种常用的抗栓剂,饭后服用课提高吸收。实验对象分为服用抗酸剂、空腹、进食三组。与空腹组相比,进食油腻食物后半小时后用药,血药浓度提高20%,而服用抗酸剂组的血药浓度恰好降低了同样程度。空腹组用药后出现胃肠道的副作用,而进食时用药则可减轻此副作用,从而增加病人服药的顺应性。
引起胃肠道刺激的药物,例如阿司匹林,应该饭时服用,因为食物可以延长胃排空时间从而时肠溶阿司匹林片的吸收增加数小时。服用阿司匹林的颗粒剂和肠溶微丸剂时延迟作用减小。因此要告知患者饭时用药所可能带来的对药效的延迟。
红霉素建议空腹服用,尽管实际上由此导致一些病人的呕吐。食物可以减轻这些症状。及时调整给药是必要的。口服大环内酯抗生素地红霉素的吸收不会因食物以及抗酸剂降低反而稍微增加。
一些药物会因特殊的治疗目的建议饭时服用。例如洛伐他丁,抑制体内胆固醇的合成,降低血液中胆固醇的浓度。
抑制胃酸分泌的药物制剂应饭前服用,以期望食物对胃酸分泌的刺激作用,例如法莫替丁和西米替丁。西米替丁建议不与抗酸剂同时使用以避免其吸收的降低。补铁剂和非甾体类抗炎药(NSAIDS)如布洛芬饭时服用以减少对胃的刺激。
用药时间不能紧紧以吸收和起效信息胃依据,一些药物给药时间不同,其药效也不同。
普伐它丁是一种治疗高胆固醇血症的药物,通常一天两次或一次,令人关注的是每天夜间服用比在早晨府哦女冠更有效。推测这与肝脏合成胆固醇在晚上12点和凌晨3点之间最多有关。对一些抗癌药来说,给药时间要求严格,因为不同的用药时间药效有着显著的变化。
大多数药物口服需要喝一杯水(大约8液量盎司),以保证药物被冲下食管。去多药物溶解有限,需要足够的液体使其溶出。一些病人数月频繁的同时使用几种药物,以至他们经常呕吐而不愿意喝大量的水。例如携带有活性HIV病毒的艾滋病患者可能用AZT或DDI以一种或多种蛋白抑制剂联合用药,这种蛋白抑制剂有沙喹那韦、印地那韦、利托那韦。这种治疗方案显示优于以前的方案,但这要看病人的顺应性,是否能够在数周内那天服用12~15粒药片。任何影响药物吸收的不顺应性都会干扰治疗效果。对于一些抗生素没有被吸收的药物可能影响胃肠道菌群。对一些引起胃肠道紊乱的药物,胃肠道残余药物很可能会加剧腹泻。
3.3.2 疾病状态对药物吸收的影响
理论上,任何引起以下几种变化的疾病都可能影响药物的吸收:(1)小肠血流(2)胃肠道的蠕动(3)胃排空时间(4)影响药物溶解度的胃部pH值(5)影响药物解离程度的小肠pH值(6)肠壁的透过性(7)胆汁的分泌(8)消化酶的分泌(9)胃肠道的菌群。一些因素可能会是主要的,而其他一些因素有可能相互抵消。有必要进行患病受试者与健康受试者的药动学研究,以衡量疾病对药物吸收的影响。
帕金森病晚期患者可能吞咽困难、胃肠道蠕动减慢。曾报道有一病例,患者口服左旋多巴因为吸收差而无法控制病情,改为静脉注射左旋多巴片剂的溶液(用J形管),可控制其症状。这种病人只能采取此治疗方案。
使用三环类抗抑郁药(阿米替林、去甲替林)和抗精神病药物(酚噻嗪)的患者反副交感神经生理作用的副作用,使胃肠道蠕动变慢,甚至造成肠梗塞。药物吸收的延迟,铁别是对缓释制剂时有发生。
胃酸缺乏病患者的胃酸分泌量不足,而胃酸对碱性药物的溶解至关重要,许多弱碱性药物因为胃中酸度不够而不能形成可溶性的盐而不溶解,由于药物碱度太弱而容易析出沉淀成游离碱的形式。
3.3.3节段性回肠炎
节段性回肠炎是末端小肠和结肠的炎症疾病,这种病会出现局部肠壁加厚、厌氧菌的过度繁殖,有时会出现肠梗阻和肠壁坏死。这对药物吸收的影响是不可预知的。可能会由于吸收面积的减小和肠壁的加厚使吸收变差。该病患者口服普萘洛尔会出现高的血药浓度,因为该病患者的体内糖蛋白水平变高影响了药物与蛋白的结合和分布的原因。
腹腔炎是近端小肠的炎症疾病,是由在谷类食物中发现的一种粘蛋白-谷蛋白的致敏性引起的。此病患者一般胃排空加快,小肠壁透过能力增强,头孢氨苄的吸收会因此提高,但不能由此作出普遍性的预测。其他会影响小肠吸收的情况包括:消化性溃疡、胃窦切除术、胃十二指肠吻合术、迷走神经切断术。
3.3.4 药物之间吸收的相互影响
溴丙胺太林是一种抗副交感神经作用的药物,可以减缓胃排空和小肠的蠕动。抗副交感神经作用的药物一般能减少胃酸的分泌。胃排空减缓是药物吸收延迟。三环类抗抑郁药和酚噻嗪也有抗副交感神经的副作用,降低胃肠道的蠕动。
甲氧氯普胺刺激胃收缩、松弛幽门括约肌、提高小肠的蠕动,从而减少一些药物的有效吸收时间,降低峰浓度和药时曲线下面积。例如:地高辛片剂的吸收可被甲氧氯普胺降低,可被抗副交感神经作用的药物如溴丙胺太林增强。能够让片剂在胃中停留较长时间溶解,有助于难溶性药物的溶出剂吸收,但对于在胃中部溶解的药物没有作用。
考来烯胺是一种不被吸收的离子交换树脂,用来治疗高血脂症。它能像活性炭一样吸附华法林、甲状腺素、洛哌丁胺等药物,减少这些药物的吸收。
钙的吸收是与维生素D协同的主动转运过程,与维生素D缺乏情况下相比,钙的吸收量高出4倍。一般认为维生素D使用后,提高钙结合蛋白的水平,从而是更多的钙从小肠中转运至血液循环中。
3.3.5影响药物吸收的营养物质因素
许多营养物质都影响药物在体内的吸收和代谢。有报道食物的摄取可提高药物的生物利用度,如普萘洛尔、美托洛尔、硝基呋喃妥英、氢氯噻嗪。但食物也可以减少许多抗生素类药物的吸收,如氨苄西林、四环素、利福平。口服药物与营养物质间的影响对不同的药物是不同的,可能增加或减少药物的吸收。一些营养物质会在药物吸收时,减少胃肠道的刺激。了解药物与营养物质间的作用可以帮助临床医生避免刺激性药物的过高血药浓度。食物还可能不改变药物的吸收而引起血药浓度的变化,例如高蛋白饮食提高药物的肝代谢,但不影响从胃肠道吸收的程度,从而引起血药浓度的降低。
水溶性维生素如维生素B12、叶酸等,具有特定的吸收机制。维生素B12与胃内的特定因子结合,形成复合物沿肠道至回肠附近,然后与特定的受体结合,维生素B12最终从复合物中解离而被吸收。
葡萄柚汁发现可以提高许多药物的血药水平,这是因为柑桔甙阻碍药物的代谢。因此血药浓度的提高不能简单的解释为吸收的增加,许多物质减少药物在胃肠道中的降解(代谢),间接的增加药物的吸收量。