企业质量管理体系
一、质量管理体系描述
1、公司于20xx年7月份通过了质量体系认证中心的质量体系认证,质量体系符合标准,并于今年6月份通过本年度质量管理体系第三方监督审核,证明了公司的质量管理体系得到了有效的实施与保持。
2、公司按标准要求制定了公司质量管理体系文件,《文件控制程序》、《记录控制程序》,各种记录记录及时、真实、完整、有效保管,便于查阅,体系总体运行情况良好。
二、管理职责
公司领导在高层领导会议上,通过宣传公司质量方针和目标,反复强调产品质量满足顾客要求与法律法规的重要性,倡导以顾客为中心,并将质量目标层层分解,落实到各部门,明确各岗位人员职责、权限,保证人员、设备、材料等方面的及时供应,确保了生产连续,质量满足规定要求。
三、资源管理
1、公司为满足顾客需求,合理组织生产,拓宽市场,不断引进人才、购买设备,以后应进一步加强对其管理,确保各种资源满足生产需要。
2、有关部门应进一步加强员工培训,以便其能力能胜任所承担的工作。
3、设备、设施管理:公司的设备、设施管理基本能按《设备、
设施管理程序》及《设备管理办法》执行。
四、产品实现
公司制定了《过程控制程序》,以加强对于生产过程中的工艺参数、人员、设备、环境等影响产品质量的所有因素加以控制。包括作业指导书,生产参数监视和设备运行状况等内容。
五、测量、分析和改进
1、公司制定了顾客满意度调查程序,通过发放调查表,电话调查,客户拜访等方法,了解产品质量和服务的满意度及对企业的整体印象。
2、在生产过程中加强对不合格产品、不合格原材料的控制,该返修的返修,该报废的坚决报废,以防止不合格的再发生。
3、各部门应有效利用《数据分析控制程序》中的适当方法,对顾客的满意、产品质量的符合性、过程特性及趋势,产品服务进行统计分析,制定各种对策,保证质量体系的良好运行。
4、公司制定了纠正预防措施控制程序,以消除不合格的原因和潜在不合格的原因,防止不合格的再发生与发生。对于体系中的不合格,各有关部门认真分析,制定切实有效的纠正、预防措施并认真实施,以便持续改进质量体系的有效性。
第二篇:医疗器械生产企业质量管理体系咨询问答专栏1
医疗器械生产企业质量管理体系咨询问答专栏
发布时间:2011-08-23
为贯彻落实国家食品药品监督管理局医疗器械质量管理体系考核相关要求,帮助企业提升对相关法规和规定的认识,促进企业实施生产质量体系管理工作,北京市药品监督管理局特建立此专栏,希望能对我市医疗器械生产企业有所帮助。
此专栏主要针对除体外诊断试剂、无菌和植入性医疗器械之外的医疗器械产品(体外诊断试剂、无菌和植入性医疗器械产品专栏另行设置)。随着国家食品药品监督管理局相关要求的变化和调整,以及新情况、新问题,北京市药品监督管理局将对此专栏内容进行相应的修改和完善。
1.医疗器械质量管理体系相关法规文件有哪些?
(1)《医疗器械注册管理办法》(国家食品药品监督管理局令第16号)
(2)《医疗器械生产企业质量管理体系考核办法》(原国家药品监督管理局令第22号)
(3)《关于规范医疗器械生产企业质量管理体系考核报告有关事宜的通知》(食药监办
[2007]145号)
(4)《北京市药品监督管理局关于印发<北京市口腔义齿监督管理的有关规定(试行)>的通知》(京药监发〔2003〕15号)
(5)《北京市药品监督管理局关于发布<北京市医疗器械软件产品监督管理规定(暂行)>的通知》(京药监械〔2006〕9号)
(6)《医用中心吸引系统、医用中心供氧系统产品技术审查规范》(京药监械〔2009〕49号)
2.医疗器械质量管理体系有效证明文件有哪些?
医疗器械生产企业根据产品类型不同,可以分别提供以下质量管理体系的有效证明文件:
(1)除体外诊断试剂、无菌和植入性医疗器械之外的医疗器械产品—北京市药品监督管理局出具的《医疗器械质量管理体系考核报告》或者由第三方颁发的医疗器械质量体系认证证书;
(2)无菌和植入性医疗器械产品—北京市药品监督管理局或国家局药品认证管理中心出具的《医疗器械生产质量管理规范检查结果通知书》;
(3)体外诊断试剂产品—北京市药品监督管理局或国家局药品认证管理中心出具的《体外诊断试剂生产企业质量管理体系考核报告》、《体外诊断试剂研制情况核查报告表》和《体
外诊断试剂质量管理体系考核有效覆盖认定表》。
3.医疗器械生产企业如何申请和咨询医疗器械质量管理体系考核相关事宜?
企业可于每周一、周四到北京市药品监督管理局医疗器械技术审评中心进行申请和咨询。地址:北京市东城区光明路13号北方亿兴大厦二层东侧,联系电话:87559566。
4.医疗器械生产企业申请医疗器械质量管理体系考核时,应提交哪些资料? 生产企业申请材料应规范清晰,签字盖章齐全,且满足以下要求:
(1)医疗器械生产企业质量管理体系考核申请书及自查表(两份,填表说明)。
(2)《医疗器械生产企业许可证》(副本)复印件,(一份);
(3)质量手册及程序文件,应符合YY/T0287-2003的要求;
(4)申请产品综述资料,至少应包括产品简介、工作原理或机理、结构及组成、生产工艺(标明关键工序、特殊过程和检验点);
(5)生产场地总平面布置图;
(6)产品注册检测报告复印件(对于产品、产品标准和说明书均没有变化的重新注册,可不提交注册检测报告);
(7)如重新注册,还应提供医疗器械产品注册证及登记表复印件;
(8)如企业通过YY/T0287质量管理体系的认证,还应提供认证证书和审核报告;
(9)如企业申请质量管理体系考核的复审,还应提交复审申请和整改报告。
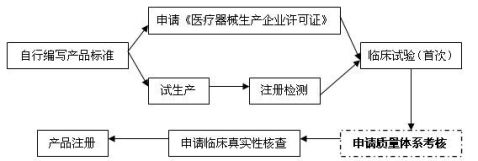
5.生产企业应在产品注册流程的哪个阶段申请质量管理体系考核?
质量管理体系考核在医疗器械注册流程中的位置如下图所示(以第三类产品首次注册为例):
6.对于进行重新注册的产品,医疗器械生产企业申请质量管理体系考核时是否需要提交注册检测报告?
根据《关于印发进一步加强和规范医疗器械注册管理暂行规定的通知》(国食药监械
[2008]409号)的规定,对于产品、产品标准和说明书均没有变化的重新注册项目,生产企业应当提交没有变化的声明,不再提交注册产品注册检测报告。
7.进行质量管理体系考核时,如何对第三类医疗器械产品注册型式检验样品进行真实性核查?
根据《北京市药品监督管理局关于开展第三类医疗器械注册核查工作有关意见的通知》(京药监械[2008]11号)规定,在对第三类医疗器械产品进行质量管理体系考核或质量管理体系认证时,现场审核组应对企业产品注册型式检验样品的真实性进行核查。
8.接受质量管理体系现场检查前,生产企业应做好哪些准备工作?
(1)应保持联系人的联系电话畅通;
(2)确保企业负责人、管理者代表、生产、质量、技术、采购等相关部门负责人及工作人员参加现场考核;
(3)应提前准备现场考核的文件及记录。一般应为检查组准备2~3套受控的质量手册、程序文件及相关工作制度、工艺规程、作业指导书等;并确保在考核现场提供体系运行的全部记录,如研发、生产、采购、检验、内审、管理评审、顾客反馈等;
(4)应准备企业基本情况、产品情况的介绍。一般应制作幻灯片,时间控制在10分钟左右。介绍材料应包括企业的基本情况、质量管理体系的建立和运行情况、考核产品的工作原理、工艺流程、产品注册情况等内容。如为整改后复审的现场检查,则应着重针对上次质量管理体系考核时检查组提出的问题,企业所采取的整改措施及完成情况等内容。
9.还有哪些应该注意的问题?
(1)企业的质量手册及程序文件首次发布和换版后,应运行质量管理体系至少三个月以上,并保存相关记录。变更生产地址的企业还应在新生产地址有效运行质量管理体系,并保存相关记录;
企业应完成内审工作,确认质量管理体系已经有效运行,再行申请质量管理体系考核。
(2)企业在申请医疗器械质量管理体系考核前,应根据相关法律法规的要求进行自查。根据自查情况,如实填写《医疗器械生产企业质量质量管理体系考核申请书》。
(3)如涉及生产地址变更或生产范围变更,应先办理《医疗器械生产企业许可证》的相关变更后,再申请医疗器械质量管理体系考核。
(4)生产企业在质量管理体系考核时应提供送检批次的产品原材料采购、生产、检验等记录。
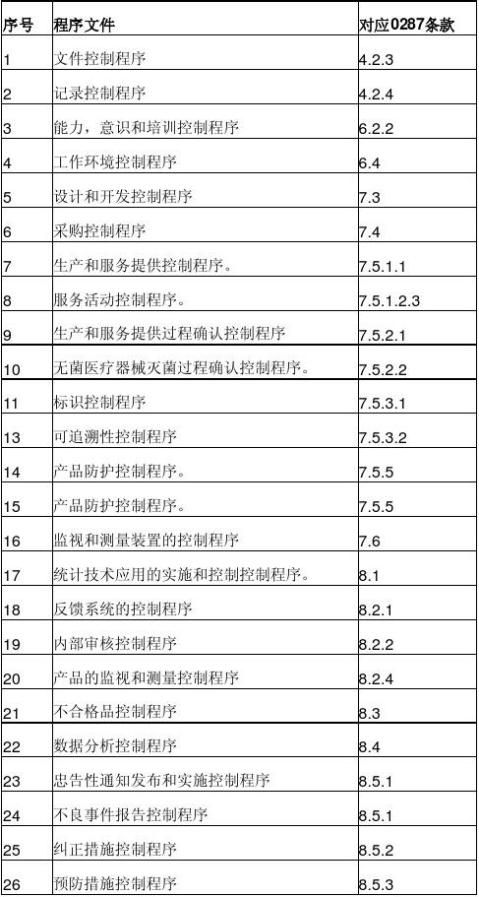
10.医疗器械生产企业质量管理体系一般应形成哪些程序文件?
企业应根据YY/T0287-2003的要求, 并结合企业和产品的具体情况建立适宜的质量管理体系,至少应包括以下程序文件: