分子生物学实验报告
实验名称:DNA克隆
一、 实验目的:
1. 学习分子生物学中最基本的技术--分子克隆(Molecular cloning,gene cloning)的操作过程。
2. 掌握基因克隆的概念;
3. 熟悉基因克隆的过程;
4. 掌握基因克隆常用仪器设备的使用方法;
5. 掌握PCR的原理;
6. 了解DNA克隆的原理及蓝白斑筛选过程的原理
7. 了解基因克隆的应用。
二、 实验原理:
1. 基本概念:
1) 基因克隆(gene cloning)或分子克隆,又称为重组DNA技术,是应用酶学方法,在体外将不同来源的DNA分子通过酶切、连接等操作重新组装成杂合分子,并使之在适当的宿主细胞中进行扩增,形成大量的子代DNA分子的过程。
2) 目的基因:准备要分离、改造、扩增或表达的基因。
3) 感受态细胞:在人工构建的质粒载体中,一般缺乏此种转移所必需的mob基因,质粒不能自行完成从一个细胞到另一个细胞的接合转移。如需将质粒载体转移进受体细菌,需将对数生长期的细菌(受体细胞)经理化方法处理后,细胞膜的通透性发生暂时性改变,成为能允许外源 DNA 分子进入的细胞,称感受态细胞。
4) 质粒是染色体外能够进行自主复制的遗传单位,包括真核生物的细胞器和细菌细胞中染色体以外的脱氧核糖核酸(DNA)分子。现在专指细菌、酵母菌和放线菌等生物中染色体以外的DNA分子。目前,已发现有质粒的细菌有几百种,绝大多数的细菌质粒都是闭合环状DNA分子(简称ccDNA)。相对分子质量一般在1Kb-200Kb之间。在基因工程中,常用人工构建的质粒作为载体。人工构建的质粒可以集多种有用的特征于一体,如含多个酶切位点、抗生素耐药性等。
5) 限制性内切酶是一种工具酶,其特点是具有能够识别双链DNA 分子上的特异核苷酸序列的能力,能在这个特异性核苷酸序列内,切断DNA 双链,形成一定长度的DNA 片段。
2. 实验方法:
1) 多聚酶链式反应是DNA的体外酶促扩增。是利用两个短的单链引物,在体外快速扩增特异DNA片段的技术。应用热稳定的聚合酶,通过双链DNA模板的热变性、引物退火和引物延伸的重复循环,使目的DNA片段在一定时间以指数方式增加百万倍。高温加热可以使 DNA双螺旋的氢键断裂,双链解离,形成单链DNA,这称为 DNA的变性。解除变性的条件后, 变性的单链可以重新结合起来,形成双链,其原有的特性和活性可以恢复,这称DNA复性, 也叫退火。引物的序列及其与模板结合的特异性是决定PCR反应结果的关键。引物设计的最大原则是最大限度地提高扩增效率和特异性;同时尽可能减少非特异性扩增。引物设计基本原则:
a) 序列应位于高度保守区,与非扩增区无同源序列。
b) 引物长度以15-30 nt为宜,一般25nt。这样长度的寡核苷酸在聚合反应温度下不会形成稳定的复合物,更短的引物一般会降低扩增特异性;
c) 碱基尽可能随机分布,G+C占45-50%。两条引物G+C含量应尽量相似;
d) 引物内部避免形成二级结构,尤其是发夹结构
e) 两引物间避免有互补序列,特别是在引物3’端,即使无法避免,其互补碱基也不应大于2个碱基,否则容易生成引物二聚体或引物二倍体;
f) 引物3’端为关键碱基;5’端无严格限制。
2) 无细胞分子克隆:在微量离心管中,加入适量的缓冲液, 微量的模板DNA,四种脱氧单核苷酸,耐热性多聚酶, 一对合成DNA的引物,通过高温变性、低温退火和中温延伸三个阶段为一个循环,每一次循环使特异区段的基因拷贝数放大一倍,一般样品是经过30次循环,最终使基因放大了数百万倍; 扩增了特异区段的DNA带。
3) 试剂盒回收目的基因:采用凝胶裂解液(如含有降低熔点的NaI)融化凝胶释放DNA后,采用特殊的硅胶树脂吸附DNA后,用洗液洗去杂质,最后用洗脱液洗出DNA。
4) 目的片段与载体连接:
a) DNA片段之间的连接主要是在T4 DNA连接酶的作用下,使DNA裂口上核苷酸裸露的3’羟基和5’磷酸之间形成共价结合的磷酸二酯键,使原来断开的DNA裂口连接起来,需ATP。通常两种不同的DNA分子可以经过下述的方法之一进行连接:
A) 同种限制酶作用不同DNA片段产生的相同粘性末端
B) 不同种限制酶作用于不同DNA片段产生的同尾末端
C) PCR产物与T载体的连接
D) 限制酶和其他方式产生的平末端。
b) T-A克隆:TaqDNA聚合酶能在平端双链DNA的3‘末端加一个碱基A。据此,在质粒DNA 3’端突出一个T来克隆PCR产物,其克隆效率比平端的连接至少高出 100倍。应用于PCR产物克隆,代表商品化载体有pCRTMⅡ,pGEM-T。
5) 转化方法(热休克法):当细胞处于 0 ~ 4 ℃, CaCl2 低渗溶液中时,大肠杆菌细胞膨胀成球状。转化混合物中的 DNA 形成抗 DNA 酶的羟基 - 钙磷酸复合物粘附于细胞表面,经 42 ℃ 数秒热激处理,促进细胞吸收 DNA 混合物。将细菌放置在非选择性培养基中保温一段时间,促使在转化过程中获得的新的表型,如氨苄青霉素耐药( Amp r )得到表达,然后将此细菌培养物涂在含 Amp 的选择性培养基上,倒置培养过夜,即可获得细菌菌落。
6) LacZ蓝白斑筛选原理:Ampicillin 抗性和 lacZ的a肽互补(蓝白斑)相结合。无质粒的受体菌,无菌斑生长;质粒转化的受体菌,有蓝色菌斑生;带外源DNA插入的质粒转化的受体菌,有白色菌斑生长。
a) b-半乳糖苷酶和Xgal会发生显色反应:b-半乳糖苷酶能把无色的化合物Xgal分解成半乳糖和一个深蓝色的物质5-溴-4-氯靛蓝。半乳糖和5-溴-4-氯靛蓝lacZ的a肽互补。(pUC质粒载体上的lacZ’ 编码a肽与这个缺失突变的b-半乳糖苷酶“互补”,使它能形成4聚体。又能分解Xgal。产生蓝色物质。
b)?插入外源基因后由于a肽互补失活,不产生蓝色反应。pUC a互补的插入失活载体上LacZ’的5‘端有一段多克隆位点(MCS)区,本身虽不干扰LacZ’的合成,但插入外源基因就会阻止LacZ’的合成。不能互补。
7) 质粒DNA提取包括三个基本步骤:培养细菌扩增质粒;收集和裂解细菌;分离和纯化质粒DNA:
a) 细菌培养物的生长:从琼脂平板上挑取一个单菌落,接种到培养物中(在含有相应抗生素的液体培养基中生长),然后从中纯化质粒,质粒的提纯几乎都是如此。现在使用的许多质粒载体(如pUC系列)都能复制得到很高的拷贝数,只要将培养物放在标准LB 培养基中生长到对数生长期晚期,就可以大量提纯质粒。
b) 细菌的收获和裂解:细菌的收获可通过离心来进行,而细菌的裂解则可采用多种方法,这些方法包括用非离子型或离子型去污剂、有机溶剂或碱进行处理及用加热处理等。具体选择哪一种方法取决于3个因素:质粒的大小、大肠杆菌菌株及裂解后用于纯化质粒DNA的技术。
c) 质粒DNA的分离纯化:所有常使用的纯化方法都利用了质粒DNA 相对较小及共价闭合环状这样两个性质。常用方法有:异丙醇沉淀;柱吸附; 氯化铯密度梯度离心。
8) 碱抽提法提取质粒DNA:将细菌悬浮液暴露于强碱性溶液中,会使细胞壁破裂,将质粒DNA释放到上清液中。当pH值恢复到中性时,它重新形成完全天然地超螺旋结构。在裂解过程中,细菌蛋白质、破裂的细胞壁和变性的染色体DNA会相互缠绕成大型复合物,与SDS一起沉淀。然后离心除去变性剂,可从上清液中回收复性的质粒DNA。
9) 质粒DNA的琼脂糖凝胶检测:DNA分子在琼脂糖凝胶中时有电荷效应和分子筛效应。DNA分子在高于等电点的溶液中带负电荷,在电场中向正极移动。在一定的电场强度下, DNA分子的迁移速度取决于分子筛效应.具有不同的相对分子质量的DNA片段泳动速度不一样,可进行分离.凝胶电泳不仅可分离不同分子质量的DNA, 也可以分离相对分子质量相同,但构型不同的DNA分子。
10) 酶切反应:反应系统包括:DNA,酶,buffer,灭菌水。限制性内切酶可以特异性的切断DNA 双链,形成一定长度的DNA 片段。限制性内切酶对环状质粒DNA有多少切口,就能产生多少酶切片段,因此鉴定酶切后的片段在电泳凝胶的区带数,就可以推断酶切口的数目,从片段的迁移率可以大致判断酶切片段大小的差别。用已知分子量的线状DNA 为对照,通过电泳迁移率的比较,就可以粗略推测分子形状相同的未知DNA的分子量。准备反应系统时应注意:
a) 不同公司的酶和buffer系统是不同的,所以一个反应应该用同一个公司的酶和buffer。
b) 酶量并不是越大越好,酶体积不应该超过总反应体积的1/10。(因为酶是储存在50%甘油中的,而反应体积中甘油的浓度超过5%就会使酶出现星号活性,即切割不应该切的位置。
c) 不同的酶配有不同的buffer,产品目录和酶的使用说明中会说明该酶用哪种buffer,有些buffer中需要另外添加BSA。
d) 重组质粒样品中不应该有酚,氯仿,酒精,EDTA,盐离子等的污染,以免影响酶的活性。
e) 反应体积,通常1到几微克DNA的反应体积可以控制在50到100微升,用20单位的酶来切。
f) 反应条件,大多数酶都是37度半小时以上。
三、实验内容:
1. 目的基因的获取(来源)
2. 目的基因与载体的连接
3. 重组DNA的转化
4. 阳性克隆的挑选与鉴定
四、实验用品
1.实验仪器:超净工作台、恒温摇床、恒温培养箱、培养皿、台式高速冷冻离心机、电泳仪,水浴锅,紫外检测仪 、1.5mL的eppendorf管、电泳槽、离心管、收集管、吸附柱、微量加样器
2.实验试剂:
1) 目的基因的获取:
a) 模板:模板DNA可以使单链分子,也可以是双链分子,不能混有蛋白酶、核酸酶、DNA聚合酶抑制剂、DNA结合蛋白类,一般100ng DNA模板/100?L。
b) 引物:浓度为0.1-0.5 ?mol/L
c) Taq DNA聚合酶:0.5-2.5 U/微升
d) dNTP:dNTP提供PCR反应的原料和能量,浓度取决于扩增片段的长度,四种dNTP浓度应相等。
e) Mg2+:Mg2+是DNA聚合酶的激活剂,其浓度对Taq DNA聚合酶影响很大,0.5mmol/L-2.5mmol/L反应体系。
2) 目的基因与载体的连接:小量DNA片段快速胶回收试剂盒、T载体 (pGEMT)、10xT4 buffer、T4 ligase
3) 重组DNA的转化:
a) LB液体培养基:称取蛋白胨(Tryptone)10 g,酵母提取物(Yeast extract) 5 g,NaCl 10 g,溶于800ml去离子水中,用 NaOH 调pH至7.4,加去离子水至总体积1L,高压下蒸气灭菌20min。
b) LB固体培养基:液体培养基中每升加15g琼脂粉,高压灭菌,倒平板。
c) 0.1mol/L CaCl2溶液:称取1.1g CaCl2(无水,分析纯),溶于90mL重蒸水中,定容至100mL,0.22µm滤器过滤除菌,4℃保存。
d) 50 mg/ml氨苄青霉素。
4) 阳性克隆的挑选与鉴定:
a) 重悬液:50mmol/L葡萄糖,25mmol/L Tris.Cl(pH8.0),10mmol/LEDTA(pH8.0)
b) 裂解液:0.2mol/L NaOH(临用前用10mol/L贮存液现用现稀释),1% SDS
c) 中和液:5mol/L乙酸钾 60ml,冰乙酸 11.5ml,水 28.5ml
d) 异丙醇 ,无水乙醇
e) 限制性内切酶EcoR I及缓冲液H ;
f) 1×TBE 缓冲液:称取Tris 10.88g、硼酸5.52g 和 EDTA 0.72g,用蒸馏水溶解后,定容至200mL,用前稀释10 倍。
g) EB 染色液:终浓度为0.5μg/mL。
 五、实验步骤:目的基因的获取 目的基因与载体的连接 重组DNA的转化 阳性克隆的挑选与鉴定
五、实验步骤:目的基因的获取 目的基因与载体的连接 重组DNA的转化 阳性克隆的挑选与鉴定
1. 目的基因的获取:
a) 变性:使双链DNA解链为单链,94摄氏度 20-30秒
b) 退火:温度由引物长度和GC含量决定,58摄氏度 30秒
c) 延伸:延伸时间由扩增片段长度决定,72摄氏度 45秒
d) 循环次数:主要取决于模版DNA的浓度,一般为25-30次
2. 目的基因与载体的连接:
1) 目的片段的胶回收
a) 将目地基因片段切下的胶转移到预先称重的1.5ml eppendorf 管中,称取重量,按照100mg=100ul的体积,加入3倍体积的溶胶液;
b) 50度水浴10min至凝胶完全溶解,每2min震荡混合;
c) 取回收试剂盒中收集柱装在收集管上,将溶解的DNA溶胶700ul转移到柱子中(大于700ul分2次),室温放置2min,12000rpm离心1min,弃去废液,将柱子套上;
d) 加入600ul 漂洗液 WB(乙醇已溶解),12000 rpm离心60s,弃去废液;
e) 加入600ul 漂洗液WB,12000rpm离心60s,弃去废液;
f) 12000rpm离心2min,甩干柱子中残液,晾干;
g) 将柱子转移到1个干净的1.5ml eppendorf 管中,加入40ul 洗脱缓冲液TE(预先水浴加热)在膜中央,室温放置2min, 12000rpm离心1min,收集离心液重新加入吸附柱,12000 rpm离心1min,收集液为洗脱的DNA;
h) 电泳检测回收效果(5ul回收片段+2ul溴酚蓝)
2) 目的片段与载体连接
a) 取1.5mL eppendorf管,对其进行编号;
b) 按下表向eppendorf管中添加试剂
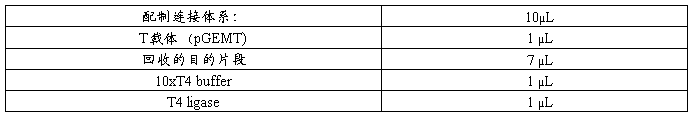
c) 16℃连接过夜;
d) 次日做转化实验
3. 重组DNA的转化:
1) 感受态细胞的制备-CaCl2法菌株活化:
a) 用接种环直接取冻存的大肠杆菌DH5α,在LB培养基平板表面划线,于37℃培养16h(过夜)。
b) 挑取一个单菌落,转到3-5mlLB培养基中,于37℃培养过夜。
c) 将培养液全部转到100mlLB培养基中(1:50—1:100放大)培养2-3小时,到OD600=0.3-0.4,使其处于对数生长期的初期。
d) 取两支50ml无菌离心管,倒入40ml培养好的菌液。冰上放置10-15min。
e) 离心10min ( 3500r/min, 4℃)。
f) 弃去上清,加入10ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮,在冰上放置30 min。
g) 离心10min ( 3500r/min, 4℃) 。
h) 弃去上清,加入4ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮。
i) 加冷冻保护剂(灭菌甘油,7V菌液:3V甘油),混匀,然后100-200μL每管分装后,超低温冰箱(-70℃)保存,有效期4-6月。
2) 感受态细胞的转化:
a) 从超低温冰箱中取100μl感受态细胞,置于冰上5min。
b) 将连接实验得到的重组质粒(10μl)加入大肠杆菌感受态细胞中,混匀,于冰上放置10-20min,使其附着。
c) 42℃水浴热击45S。
d) 热击后迅速置于冰上放置3min。
e) 加入1ml LB液体培养基于37 ℃培养1-1.5h(低转速,150-180r/min)。
f) 10000rpm离心1min,吸去上清,剩余约200ul。
g) 加入IPTG 16 μl(100mg/ml),X-Gal 40 μl(40mg/ml)。混匀,涂布在含Amp的培养基平板上,正面向上放置半小时,待菌液完全被培养基吸收。
h) 将平板倒置于37℃恒温培养箱中培养12-16个小时。
i) 次日观察结果,挑克隆培养。
3) 热休克法获取细菌菌落:
a) 准备:100微升感受态菌+10微升载体DNA。
b) 吸附DNA:置于冰上混合,静置十分钟。
c) 摄入DNA:温度为42摄氏度,时间45秒。
d) 加入一毫升LB培养基。
e) 37摄氏度下,置于恒温摇床一小时。
f) 200微升转化液+IPTG+X-Gal涂含抗菌素的平板。
4. 阳性克隆的挑选与鉴定:
1) 质粒提取:
a) 柱平衡:吸附柱放入收集管,向吸附柱CP3中加入500ul平衡液BL,12000rpm离心1min,倒掉废液,备用。
b) 取2ml过夜培养的菌液加入1.5ml eppendorf 离心管中,12000rpm离心一分钟,弃去上清。
c) 向留有菌体的离心管中加入250ul溶液P1(预先加入RaseA),震荡混匀,使菌体悬浮。
d) 想离心管中加入250ul溶液P2,温和上下翻转8次使菌体充分裂解。此时菌液清亮粘稠,所用时间不超过5分钟
e) 向离心管中加入350ul溶液P3,立即温和上下翻转8次,充分混匀。此时溶液中出现白色絮状沉淀。12000rpm离心10min。
f) 将上清液转移入平衡好的吸附柱CP3中,注意不要吸出沉淀,12000rpm离心1min。倒掉废液,将吸附柱放回收集管。
g) 向吸附柱中加入600ul漂洗液PW(预先加入无水乙醇),12000rpm离心1min,弃去废液。
h) 重复第7步。
i) 再次离心12000rpm,2min.
j) 将吸附柱放入干净1.5ml eppendorf管中,向吸附膜中间滴加50-100ul洗脱缓冲液EB,室温放置2min,12000rpm离心2min收集质粒。
k) 琼脂糖凝胶电泳
2) 酶切鉴定:
a) 反应体系的建立: (20μl体系):H2O12μl、质粒DNA(200 ng/μl) 5 μl、10×酶切缓冲液2μl、EcoRⅠ(10 U/μl) 1μl用离心机在12000 rpm下离心10秒
b) 37 ℃水浴1.5 h-2h或4℃过夜。
c) 将Eppendorf管置65 ℃水浴中10 min,通过加热使酶失活以终止反应,或加入2ul loading buffer终止反应。
d) DNA 琼脂糖凝胶电泳检测
六、实验注意事项
1. 目的基因的获取
1) 加样时在超净工作台操作,先加对照组模板,在加实验组模板;
2) 防止引物中污染进模板,将引物分装使用;
3) 用于PCR的试剂小量分装使用;
4) 设置阳性和阴性对照,重复实验;
2. 目的基因与载体的连接:
1) 连接缓冲液的影响:
a) 20-100mmol/L的Tris-HCl,较多用50mmol/L,pH的范围在7.4-7.8,目的是提供合适酸碱度的连接体系;
b) 10mmol/L的MgCl2,作用是激活酶反应;
c) 1-20mmol/L的DTT,较多用10mmol/L,作用是维持还原性环境,稳定酶活性;
d) 25-50ug/ml的BSA,作用是增加蛋白质的浓度,防止因蛋白浓度过稀而造成酶的失活。
e) 0.5-4mmol/L的ATP,现多用1mmol/L,是酶反应所必需的。
注:含有ATP的缓冲液应于-20℃保存,溶化取用后立即放回。连接缓冲液体积较大时最好分小管贮存,防止反复冻融引起ATP分解。
2) 连接温度与时间的影响:因为黏性末端的DNA双链间有氢键的作用,所以温度过高会使氢键不稳定,传统上将连接温度定为16℃,时间为4-16h。现经实验发现,对于一般的黏性末端来说,低温连接较长的时间可取得较好的连接效果。
3) 酶浓度的影响:日常使用的DNA浓度比酶单位定义状态低10-20倍,连接平末端时酶用量要比连接黏端大10-100倍。
4) DNA浓度的影响:要求得到环化的有效连接产物, DNA浓度不可过高,一般不会超过20nmol/L。要求线性化的连接产物, DNA的浓度可以高些,至少是接近推荐的浓度。
3. 重组DNA的转化:
1) 注意细菌的生长状态:实验中应密切注视细菌的生长状态和密度,尽量使用对数生长初期的细胞(一般通过检测OD600来控制。DH5?菌株OD600为0.5时细胞密度是5×107/ml)。
2) 注意操作的条件环境:所有操作均应在无菌条件和冰上进行。感受态细胞长时间处在常温状态下,会严重影响到其转化率,因此应尽量减少常温操作,动作要迅速。
3) 注意操作的力度:为减少对细胞的机械损伤,操作时动作不能剧烈。
4) 注意保持所使用器皿的洁净性:迹量的去污剂或其它化学物质的存在可能大大降低细菌的转化效率。
5) 注意化合物及无机离子的影响:在Ca2+的基础上联合其他二价金属离子(如Mn 2+或Co 2+)、DMSO或还原剂等物质处理细菌,可使转化效率大大提高(100-1000倍)。
4. 阳性克隆的挑选与鉴定:
1) 吸样量一定要准确;
2) 为了不使酶污染而导致浪费,最后才加酶;
3) 要求在冰上操作,并充分混匀;
4) 开启Eppendorf管时,手不要接触到管盖内面,以防污染;
5) 样品在37 ℃与65 ℃保温时,将离心管盖严,以防水进入管内造成实验失败。
七、实验结果
八、实验思考
基因克隆完整实验过程如下:
一.目的基因的获取。DNA克隆的第一步是获得包含目的基因在内的一段DNA分子,该段目的基因可以直接来自DNA并由切割产生,也可来自mRNA并由逆转录酶逆转录成cDNA获得。当然,在NCBI上获取目的基因的序列后可直接交付给公司合成。由于初始获得的目的基因水平在ng级,故需要在体外使其迅速扩增,最理想的方法是PCR仪自动化扩增。PCR的基本过程是在试管内加入含有待扩增DNA片段的双链DNA,分别能与待扩增DNA片段两侧的特定序列互补的两个寡核苷酸引物,4中dNTP,含有一定弄得Mg2+的缓冲液,和耐热的DNA聚合酶,通过加热到94℃左右使DNA变形,降温到55℃左右使引物与模板结合,升温到72℃左右合成新链的3个步骤循环。由此目的基因呈指数形式增长。
二.载体的选择做基因克隆常用的载体有:质粒载体,噬菌体载体等。从总体上讲,根据载体的使用目的,载体可以分为克隆载体,表达载体,测序载体,穿梭载体等。并且载体应具有一些基本的性质:1.在宿主细胞中有独立的复制和表达的能力,这样才能使外源重组的DNA片段得以扩增。2.分子量尽可能小,以利于在宿主细胞中有较多的拷贝,便于结合更大的外源DNA片段。3.载体分子中最好具有两个以上的容易检测的遗传标记,赋予宿主细胞的不同表型特征。4.载体本身最好具有尽可能多的限制酶单一切点,为避开外源DNA片段中限制酶位点的干扰提供更大的选择范围。
三.体外重组
体外重组即体外将目的片断和载体分子连接的过程。大多数核酸限制性内切酶能够切割DNA分子形成有粘性末端,用同一种酶或同尾酶切割适当载体的多克隆位点便可获得相同的粘性末端,粘性末端彼此退火,通过DNA连接酶的作用便可形成重组体。而当目的基因被切割成平末端后也可与载体的平末端连接,但连接效率没有粘性末端的高。当然,有时为了不同的克隆目的,如将平端DNA分子插入到带有粘末端的表达载体实现表达时,则要将平端DNA分子通过一些修饰,如同聚物加尾,加衔接物或人工接头,PCR法引入酶切位点等,可以获得相应的粘末端,然后进行连接,此为修饰粘末端连接。
四.导入受体细胞
载体DNA分子上具有能被原核宿主细胞识别的复制起始位点,因此可以在原核细胞如大肠杆菌中复制,重组载体中的目的基因随同载体一起被扩增,最终获得大量同一的重组DNA分子。原核宿主细胞一般压迫月排名高Ca2+处理使其呈感受态。呈感受态的细菌细胞膜出现变化并且容易使表面吸附的DNA进入细胞,从而达到导入目的基因的目的。当然也可以用电击法转化,该方法操作简单,转化率高,但此法需要特殊设备并且电压较高、脉冲时间长,细菌会有相当高的死亡率【2】。
五.重组子的筛选。重组子转化宿主细胞后,载体上的一些删选标志基因失活从而导致细菌的某些表现型改变,通过在琼脂板中添加一些相应的筛选物质,之一直接筛选鉴别含有重组子的菌落。现在使用的许多载体都带有一个大肠杆菌的DNA的短区段,其中有β-半乳糖苷酶基因(lacZ)的调控序列和前146个氨基酸的编码信息。在这个编码区中插入了一个多克隆位点(MCS),它并不破坏读框,但可使少数几个氨基酸插入到β-半乳糖苷酶的氨基端而不影响功能,这种载体适用于可编码β-半乳糖苷酶C端部分序列的宿主细胞。因此,宿主和质粒编码的片段虽都没有酶活性,但它们同时存在时,可形成具有酶学活性的蛋白质。
这样,lacZ基因在缺少近操纵基因区段的宿主细胞与带有完整近操纵基因区段的质粒之间实现了互补,称为α-互补。由α-互补而产生的LacZ+细菌在诱导剂IPTG的作用下,在生色底物X-Gal存在时产生蓝色菌落,因而易于识别。然而,当外源DNA插入到质粒的多克隆位点后,几乎不可避免地导致无α-互补能力的氨基端片段,使得带有重组质粒的细菌形成白色菌落。如需进一步鉴定,应将筛选出来的细菌质粒提取出来进行序列分析。
2.材料与方法。
2.1 材料 PCR仪,电泳仪,EP管,移液枪,琼脂糖凝胶,水浴锅,离心机,振荡器,平衡管等。
2.2 方法
PCR体系:
rTaq(5U/ul) 1.0ul
dNTP(2.5mM) 4.0ul
引物1(20uM) 1.0ul
引物2(20uM) 1.0ul
模板 1.0ul
Buffer(10*) 5.0ul
H2O 37.0ul
总体积 50.0ul
PCR循环条件
94℃ 3 min
94℃ 30 s
60℃ 30 s 30 cycle
72℃ 1 min
72℃ 10 min
4℃ forever
1%琼脂糖胶制备
1.准备好制胶槽,称取1.5g琼脂糖粉;
2.用165ml TAE加热溶解(因为加热溶解时会有蒸发损耗);
3.室温冷却至不太热时,加入7.5ul EB;
4.倒入制胶槽,注意尽量不要产生气泡,若有气泡,用枪吸出;
5.室温待其凝固,拔出梳子。
目的片段的胶回收
用胶回收试剂盒
1、将目标片段切下的胶转移到预先称重的1.5ml eppendorf 管中,称取重量,按照100mg=100ul的体积,加入3倍体积的溶胶液DD;
2、56度水浴10min至凝胶完全溶解,每2min震荡混合;
3、取回收试剂盒中收集柱装在收集管上,将溶解的DNA溶胶700ul转移到柱子中(大于700ul分2次),室温放
置1min,12000rpm离心1min,弃去废液,将柱子套回;
4、加入700ul 漂洗液 WB(乙醇已溶解),12000 rpm离心30s,弃去废液;
5、加入500ul 漂洗液WB,12000rpm离心30s,弃去废液;
6、12000rpm离心2min,甩干柱子中残液;
7、将柱子转移到1个干净的1.5ml eppendorf 管中,加入50ul 洗脱缓冲液EB(预先水浴加热)在膜中央,室温放置2min, 12000rpm离心
1min,收集离心液重新加入吸附柱,12000 rpm离心1min,收集液为洗脱的DNA;
电泳检测回收效果
连接载体
步骤:
1、取0.2mL ep管,编号;
2、配制连接体系:10μL
1 μL T载体
3 μL 目的片段
5 μL Solution I
1 μL 无菌水
3、16℃连接2h;
感受态细胞的制备
1.用接种环直接取冻存的大肠杆菌DH5α,在LB培养基平板表面划线,于37℃培养16h;
2.挑取一个单菌落,转到3-5mlLB培养基中,于37℃培养过夜;
3.将培养液全部转到100mlLB培养基中(1:50—1:100放大)培养2-3小时,到OD600=0.3-0.4,使其处于对数生长期的初期;
4 .取两支50ml无菌离心管,倒入40ml培养好的 菌液,冰上放置10-15min;
5 .4℃下3500r/min 离心10min(4℃);
6 .弃去上清,加入10ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮,在冰上放置30 min;
7 . 4℃下3500r/min离心10min(4℃);
8 .弃去上清,加入4ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮;
9 .细胞悬浮液一般不立即用于转化实验,而是在4℃冰箱放12-16h(过夜),次日加冷冻保护剂(灭菌甘油,7V菌液:3V甘油),混匀,然后100-200μL每管分装后,超低温冰箱(-70℃)保存,有效期4-6月。
质粒DNA的转化
1. 从超低温冰箱中取100μl感受态细胞,立即置于冰上;
2. 实验组:将连接实验得到的重组质粒(含量不超过
50ng,体积5μl)加入大肠杆菌感受态细胞中,混匀,于冰上放置30min,使其附着;
设对照:质粒组(阳性)空白组(阴性)
3. 42℃水浴45s;
4. 热击后迅速于冰上放置3min;
5. 加入800μL-1ml LB液体培养基于37培养1-1.5h(低转速,150-180r/min);
6. 离心,倒去上清,剩余约200ul上清,加入16ul IPTG,50ul X-gal,吹匀;
7. 将培养液均匀涂布在含Amp+的培养基平板上,正面向上放置半小时,待菌液完全被培养基吸收;
8. 将平板倒置于37℃恒温培养箱中培养12-16个小时,然后观察结果;
质粒的提取
(1)培养细菌:大肠杆菌接种到LB培养基2ml-5ml中,37度振荡培养8-16小时;
(2)取培养液1.5ml于Eppendorf管中,12000rpm离心1分钟,去掉上清液,重复收集一次。加入300ul溶液I,充分吹打混匀后,室温放置2分钟;
(3)加入300ul新配置的溶液Ⅱ(10ul NaOH +10ul SDS,用蒸馏水稀释至1ml),轻柔颠倒5-10次;
(4)加入300ul溶液Ⅲ,颠倒5-10次,放置2分钟;
(5)12000rpm离心10分钟,取上清液(不大于700ul)于一新的Eppendorf管.加入等体积异丙醇,混匀后放置5分钟,12000rpm离心10分钟,弃去上清;
(6)用无水乙醇清洗一次,离心管倒置在吸水纸上2-3min,自然干燥;
(7)加入30ul TE 溶液溶解提取物,充分溶解;
(8)琼脂糖凝胶电泳检测提取结果;
酶切鉴定
使用限制性内切酶获得粘性末端的目的基因
4.讨论
基因克隆是一项在分子生物学领域内具有革命性的研究技术,其实验目的是是目的基因能够在一定的宿主细胞中进行大量扩增,该技术的出现将分子生物学研究大大推进,随着科技的发展,该技术已成为进入分子生物学领域内必掌握的基础实验之一。
本次实验历时一周,作者同本组成员共同完成了基因克隆实验。基因克隆实验是有一系列实验共同组成,相比于作者之前在基础部做过的实验,基因克隆实验更能提升实验人员的实验操作能力与分析问题、解决问题的能力。作者先前曾独立完成免疫组织化学实验,对免疫组织化学实验较为了解。免疫组织化学实验的关键在于冷冻切片的质量与一抗抚育的效果,而基因克隆实验中注意事项、随机因素多,更要求实验人员极为细心,并对实验有充分的了解。
如今分子生物学家已将人类基因组测序工作完成,正致力于研究基因的功能,当然要研究基因的功能必须要了解基因产物蛋白质的结构与功能,(当然也包括相对于蛋白质总量来说是少量的RNA承担着一部分生物学功能),基因序列千变万化,基因产物多姿多样。如果可以将一段基因与其翻译的蛋白质结构域最基本的对应关系找出来,然后利用结构域组装的方式来构建需要的蛋白质,这样也是一种靶向治疗的方法。但蛋白质的进化是爆炸式的,可能相近的蛋白质在结构中仅有一点点的不同,但其执行的功能是大相径庭。