分子克隆常用技术
一、核酸的纯化
在分子克隆的所有操作中,最基本的操作是核酸的纯化。其关键步骤是去蛋白质,通常只要用酚/氯仿。氯仿抽提核酸的溶液即可。每当需要把克隆有某一些所用的酶灭活或去除以便进行下一步时,可进行这种抽提。然而,如要从细胞裂解液等复杂的分子混合物中纯化核酸,则要先用某些蛋白水解酶消化大部分蛋白质后,再用有机溶剂抽提。这些广谱的蛋白酶包括链霉蛋白酶及蛋白酶K等,它们对多种天然蛋白均有活性,(1)用酚氯仿抽提:这两种有机溶剂合用,比单独用酚抽提的除蛋白效果更佳。继而用氯仿抽提则可除去核酸制品中的痕量酚。①核酸样品置有盖小离心管中,加入等体积的酚/氯仿。②旋涡混匀管内容物,使呈乳状。③12000×g室温离心15秒。④水相移入另一离心管,弃去两相界面和有机相。⑤重复步骤①-④步操作,直至两相界面上见不到蛋白质为止⑦按下述核酸浓缩法沉淀回收核酸。
二、核酸的浓缩
应用最广的核酸浓缩法是乙醇沉淀法。在中等浓度单价阳离子存在下,加入一定量的乙醇后,所形成有核酸沉淀可经离心而回收,甚至对低至pg量的DNA或RNA,也可定量回收。回收的核酸可按所需浓度,再溶于适当的缓冲液中。 具体操作时,可向含样品的小离心管中加入V/10单价阳离子盐贮存液2V无水乙醇,混匀,放冰水浴中15-30min,取出目测平衡,0-4度,12000g,离心10min。吸弃上清,再另70%乙醇0.5-1ml,12000g,0-4度洗涤离心2min。吸弃上清,沉淀用油泵抽干或打开盖子晾干后,溶于适当体积的缓冲液中。
单价阳离子盐溶液
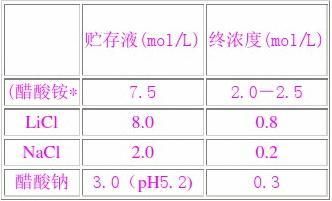
*:当加醋酸铵时,需加入DNA液的V/2。
单价阳离子盐的选择,主要基于下述考虑:用醋酸铵可减少dNTP的共沉淀,但在以后要作核酸的磷酸化时应避免用醋酸铵,因铵离子是T,多核苷酸激酶的强烈抑制剂。当用较高浓度的乙醇沉淀RNA时,常用LiCl,因LiCl在乙醇中溶解度很高,不随核酸共沉淀。含有SDS的核酸样品,应使用NaCl,这时该去垢剂
要70%乙醇中仍保持可溶。DNA和RNA沉淀,大多使用醋酸钠(pH5.2 )。
三、DNA、RNA的定量
准确的方法是紫外分光光度法。但本法要求核酸样品是纯净的(即无显著的蛋白质、酚、琼脂糖或其它核酸、核苷酸等污染物的制品)。用紫外分光光度计测定260nm和280nm两个波长处的光吸收,然后,按IA260相当于50μg/ml双链DNA。40μg/ml单链DNA或RNA及20μg/ml单链寡核苷酸。计算你的样品含量。260nm和280nm两处读数的比值(A260/A280),可反映核酸的纯度。DNA和RNA纯品的A260/A280的值分别为1.8和2.0如果样品中有蛋白质或酚的污染,则A260/A280将明显低于此值,此时就无法对样品中的核酸进行精确定量。可将样品纯化后再作定量测定。有丰富实验室经验的人,仅凭样品电泳后溴化乙锭染色萤光带的强度,即可大致判断出样品中的核酸含量,故他们常不作核酸的紫外分光光度法定量。
四、核酸的凝胶电泳和分子量参照物
(一)琼脂糖凝胶电泳
可用于分离、鉴定和提纯DNA片段。本法操作简单、迅速,能分辨其它方法不能分开的DNA片段混合物,分开的DNA可用低浓度的萤光染料(0.5μg/ml溴化乙锭)染色,在紫外灯下直接观察检测少至1ng的DNA。
DNA通过琼脂糖凝胶的迁移率取决于以下参数:①DNA分子的大小:线奖双链DNA分子通过凝胶的速率与其分子量的常用对数成反比。据此用已知分子量的标准物质和待测分子量的DNA片段同时电泳,比较其电泳速率,即可求出待测片段的分子大小。②琼脂糖浓度:给定大小的DNA片段,以不同速度通过不同浓度的琼脂扩凝胶。因此利用不同浓度的凝胶,可分辨范围广泛,大小不同的DNA片段
不同浓度琼脂糖凝胶的分离范围
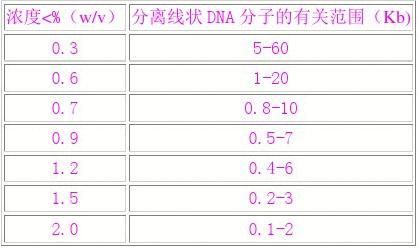
③DNA构型:相同分子量的闭环(Ⅰ型),开环(Ⅱ型)和线状(Ⅲ型)DNA,以不同速率通过凝胶,一般情况下,迁移率Ⅰ型>Ⅲ型>Ⅱ型。④应用的电压:在低电压时,线状DNA片段的迁移率与所用电压成正比。但是压增高时,大分子
量DNA片段迁移率的增大是不同的,因此琼脂糖凝胶的有效分离范围随电压增大而减小。为了获得DNA片段的最大分辨力,凝胶电泳时电压不应超过5V/cm。 (二)聚丙烯酰胺凝胶电泳
可用于分析和制备小于1Kb长度的DNA片段
DNA在聚丙烯酰胺凝胶中的有效分离范围
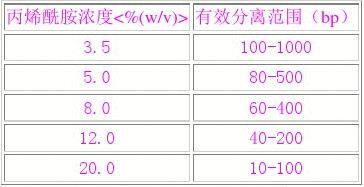
聚丙烯胺凝胶多用垂直平板电泳,准备凝胶时,先配制30%单体母液(29克丙烯酰胺,1克双丙烯酰胺,加水溶解,定容至100ml),再用它来配制所需浓度的凝胶。每100ml上述液体加30μl四甲基乙胺(TEMED),混匀后即可灌注于予先准备好的洁净不渗漏的凝胶玻板,待凝胶灌至近顶端时,立即插入合适的“梳子”,放室温聚合60分钟,若冬天室温太低,则可放37度温温箱内,以促进聚合。聚合完成后,拔出“梳子”,将凝胶板固定于电泳槽中,向电泳槽倾入1×TBE,用滴管冲洗加样孔和凝胶底部以除去气泡,即可加样电泳。一般所用电压1-8v/cm,随时观察标记染米的迁移。在溶于1×TBE的聚丙烯酰胺中,标记染料迁移速率与下述DNA片段的速率相同
标记染料在PAG中的迁移*
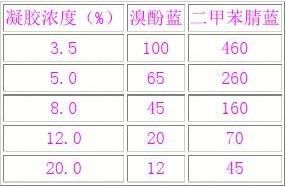
*这些数字是与染料共同迁移的DNA片段的近似大小(以bp计)。电泳结束,从电槽中取出玻板并小心地撬开,凝胶浸于溴化乙锭液(0.5μg/ml1×TBE)染色,45min后放紫外灯下观察电泳结果。
(三)分子量参照物
为了判断目的DNA片段的大小,常在同一凝胶的目的DNA旁加一分子量参照物,同时电泳并染色后,就能在紫外灯下很快知道目的片段的大小。最常用的分
子量参照物是 Hind Ⅲ消化物,各片段的大小以bp表示,分别为:23130,9416,6557,4361,2322,2027,564,125
在微型离心管中乙醇沉淀的标准方法
(1)估计DNA溶液的体积。
(2)调整单价阳离子的浓度。若DNA溶液中含有高浓度的盐,可用TE(pH8.0)
稀释,否则加入表A8-1列出的一种盐。
如果DNA溶液的体积不超过400ul可在单个微型离心管中进行沉淀,体积较大时可分成数管,或者在适宜于中速离心或超离心的离心管中进行离心。
(3)充分混匀溶液,准确加入2倍体积的冰冷乙醇,再充分混匀溶液。将此
含乙醇的溶 液置冰上使DNA沉淀形成。
一般放置15-30min就足够了。但如果DNA分子太小(小于100个核苷酸)或者DNA的含量太低(低于0.1ug/ml)则需把在冰上放置的时间延长到1h以上并加入MgCl2至终浓度0.01mol/L。DNA可在含乙醇的溶液中于0℃或-20℃无限期地
保存。
(4)于0℃离心回收DNA。
在多数情况下于最大转速离心10min就足够了。然而如以上所述,在沉淀低浓度DNA(低于20ng/ml)或非常小的片段时则需要延长离心的时间。
图A8-2 吸出上清液
手持打开盖子的微型离心管成一定角度,使沉淀物在上侧,将一个一次性吸头连至真空管道,从管内抽吸液体。把吸头置于离心管下侧,恰好在液体的凹面以下。随着液体的吸出,可将吸头移向管底。抽吸
要温和,以免沉淀吸入吸头,并应使吸头的尖部离开沉淀。最后吸尽附着于管壁的液滴。
(5)用自动微量移液器或连于真空管道的一次性吸头(见图A8-2)小心移出上清液,注 意不要扰动沉淀(有时沉淀是看不见的)。用吸头吸尽附于管壁的所有液滴。在沉淀比较珍贵的DNA样品时,最好暂时留存上清液,直至确定已回收
到沉淀的DNA后再废弃。
(6)加半管70%乙醇,于4℃以最高转速离心2min。
(7)重复第5步。
(8)将离心管于室温下敞口放置在实验台上,直至残留的液体挥干。过去常用冻干机干燥沉淀,这一步不仅没有必要,而且也不适当,因为这样会引起小片段DNA(小于 400个核苷酸)的变性(Svaren et al,1987),还大大降低大
片段DNA的收率。
(9)将DNA沉淀(沉淀常常是看不见的)重新溶解于适当体积的缓冲液(一般为
pH 7.6-8.0的TE),用缓冲液充分漂洗管壁。
[注]i在微型离心管中离心之后,并非所有DNA沉淀都沉积在管底,沉积在管壁的DNA最多可达50%。为了回收到所有DNA,应使液滴在管壁的适当部位
来回滚动,可用微量加液器上的一次性吸头推动液滴。
ii可用1倍体积的异丙醇<!>代替2倍体积的乙醇沉淀DNA。异丙醇沉淀的优点是离心液体的体积较小,但异丙醇挥发性较乙醇差而难于去除,另外有些溶质如蔗糖和氯化钠等在用异丙醇时更容易共沉淀。总之,除非必须降低液体的体
积,一般最好用乙醇沉淀。
iii一般用乙醇从溶液中沉淀出来的DNA重溶于低离子强度的缓冲液如TE(pH8.0)中比较容易,偶尔在用含MgCl2或浓度高于0.1mol/L Nacl的缓冲液直接溶解DNA沉淀时可能要遇到一点困难。因此最好是先用少量低离子强度的缓冲液将DNA溶解后再调节缓冲液的成分。若样品不易溶于较小体积的缓冲液,可用多一些缓冲液溶解,再重新用乙醇沉淀。第二次沉淀有助于去除多余的盐及可能妨
碍DNA溶解的其他成分。
用乙醇沉淀RNA
RNA可以用含2.5-3.0倍体积的乙醇从含有0.8mol/L LiCl、5mol/L醋酸铵或0.3mol/L醋酸钠的溶液中有效地沉淀下来。选择上述哪一种盐要取决于其后RNA的用途。由于十二烷基硫酸钾盐的极难溶,若要将沉淀得到的RNA溶于含SDS的缓冲液(如用寡聚dT-纤维素层析时就使用这种缓冲液)则应避免使用醋酸钾。同样,如果RNA已溶于含有SDS的缓冲液中,沉淀时也不要用醋酸钾。
[注]用于沉淀RNA的溶液应是无RNA酶的(见第7章)。
用氯化锂沉淀大分子RNA
小分子RNA(如tRNA和5SrRNA)在离子强度高的溶液中可溶,而大分子RNA(如
rRNA和mRNA)则不溶,可通过离心沉淀下来。
(1)测量样品的体积,加0.2倍体积无RNA酶的8mol/L LiCl,充分混匀,置
冰浴中至少2h。
(2)于0℃以15000g离心20min,弃上清,将沉淀下来的大分子RNA重溶于0.
2倍体积水中。
(3)重复1和2步。
(4)用2倍体积乙醇沉淀,从重悬的沉淀物中回收大分子RNA。
用微量浓缩器进行核酸的浓缩及脱盐
除乙醇沉淀外,超滤也是核酸溶液浓缩和脱盐的一种可供选择的方法。这种方法不需要相的变化,在处理低浓度核酸时特别有用。Millipore公司供有一种称为Microcon滤筒的离心超滤装置,可以有效地进行核酸的脱盐和浓缩。下述方案及加注采自Millipore公司的网站(WWW.millipore.com),详细说明可从该
网站上查找。
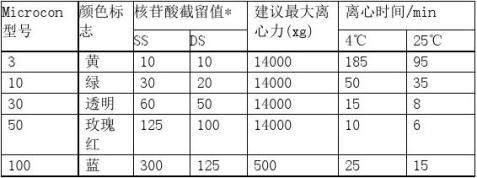
(1)选择一种型号的Microcon,其核苷酸截留值等于或小于待分离核酸的大
小(见表 A8-3)。
(2)如图A8-3所示,从提供的两只小管中取出一只,将Microcon滤筒插入
其中。
(3)浓缩时(不影响盐的浓度),可移取最多达500ul的样品(DNA或RNA)加到样品 池中,按说明书推荐的时间离心,转速不要超过表A8-3列出的离心力。
(4)若进行盐的交换,加适量适当的稀释液,使浓缩样品的体积至500ul。按说明书推 荐的时间离心,转速不要超过表A8-3列出的离心力。欲使盐的
浓度更低,必要时可重复整个步骤。见表A8-3的脚注。
重要:样品池不要加得太满。
(5)从小管上取下样品池,倒置插入另一只小管中(在分析样品之前要留存滤
液)。
(6)在一只微型离心管中于500—1000g离心2min回收小管中的核酸。
(7)取下样品池,盖紧管盖保藏样品。
图A8-3 用Microcon超离心进行核酸溶液的浓缩及脱盐
表A8-3 微型浓缩gSMic忱on的核苷酸截留值
注:注意单纯超滤是不会改变缓冲液组成的。在Micrpcon中离心浓缩样品的盐浓度与原始样品的盐
浓度相同。
脱盐时可在浓缩的样品中加水或缓冲液至原先体积,再重新离心(称作不连续渗滤)。这种脱盐的方式与超滤的浓缩系数有关。举例来说,一份含100mmol/L盐的500ul样品浓缩至25ul时(浓缩系数为20),则样品中95%的盐被去除,而样品中的盐浓度仍为100mmol/L。再将样品用水稀释至500ul时盐的浓度降至5mmol/ml,再将此稀释的样品浓缩至25弘1可去除原先总量的99%的盐,这时浓缩样品盐浓度为0.25mmol/L(译者注:原文有误,0.25mmol/L是再将此浓缩样品用水稀释至500ul后的浓度)。若欲脱盐更彻底一
些,可再进行一次溶解和离心的循环,即可脱去原先99.9%的盐量。
*ss表示单链,ds表示双链。
用丁醇抽提法浓缩核酸
在用仲丁醇(异丁醇)或正丁醇<!>抽提水溶液时,一些水分子会分配到有机相中,抽提数轮之后能够显著降低核酸溶液的体积。这个方法可降低稀核酸溶液
的体积,以致容易通过乙醇沉淀加以回收。
(1)测量核酸溶液的体积,加等体积异丁醇,用旋涡混合器混匀。 加入过多异丁醇会除去溶液中的全部水分而导致核酸沉淀。如果发生这种情况,可在有机相中加水,
直到重新出现水相(其中含有核酸)。
(2)在微型离心管中以最高转速室温离心20s,或用台式离心机1600g离心1
min。用 自动微量移液器移出并弃掉上层(异丁醇层)。
(3)重复1和2步直至水相达到所需体积。
由于异丁醇抽提并不能除盐,水溶液中的盐浓度会随溶液体积减小而成比例地增高。可通过离心柱层
析或乙醇沉淀将核酸转换到所需的缓冲液中。
第二篇:分子克隆技术实验操作手册
生物秀实验频道——生命科学实验中心
实验操作手册2005
生命科学技术学院
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心课程简介
分子克隆技术是指DNA的无性繁殖技术。分子克隆技术课程主要是针对生物技术专业、生物科学专业、生命科学与技术基地班本科生及生物学类专业研究生而开设的实验课。实验内容涉及分子克隆的一些基本方法及基本操作技巧,主要包括分子克隆和分子杂交两大部分:
分子克隆技术:DNA重组技术是分子生物学的核心内容。本实验利用质粒载体克隆外源DNA片段,通过这个实验大家可以掌握质粒载体的抽提、外源DNA的准备、酶切、连接及感受态细胞的制备、连接产物的转化以及阳性克隆子的鉴定和验证等。
分子杂交技术:实验室常用的分子杂交技术主要有Southern blotting,Northern blotting,Western blotting及Dot blotting等。Southern blotting是通过用一种或多种限制性内切酶消化基因组或其它来源的DNA,经过琼脂糖凝胶电泳按大小分离酶切所得的片段,随后DNA在原位发生变性并从凝胶转移到一固相支持物上(硝酸纤维素膜或尼龙膜)。DNA转移至固相支持物的过程中各DNA的相对位置保持不变,用一定方法(如放射性同位素)标记的DNA探针与固着在膜上的DNA 杂交,经X-光片自显影显现出与探针DNA互补的DNA电泳条带的位置,然后进行分析。本实验要求掌握植物总DNA的抽提、质量检测、限制性内切酶操作、DNA的琼脂糖凝胶电泳、转膜、探针的制备、同位素操作等方面的实验技术。在转录水平上研究和了解基因的表达与调控是分子生物学和基因操作的重要内容。为让学生初步了解有关RNA的操作过程注意事项,我们还列出Northern blotting的操作步骤以供选择。
1
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心目 录
系列一 分子克隆技术.....................................................................................................4
实验一 质粒的制备......................................................................................................4 实验二 DNA的琼脂糖凝胶电泳........................................................................5 实验三 外源DNA片段在质粒载体中的克隆..........................................6 实验四 感受态细胞的制备.....................................................................................9
CaCl2感受态细胞的制备实验步骤.......................................................................................9
电转化法制备大肠杆菌感受态细胞的实验步骤................................................................10
实验五 质粒的转化及转化子的鉴定.............................................................11
热激法转化实验步骤:........................................................................................................11
质粒电转化大肠杆菌感受态细胞操作步骤........................................................................12
实验六 PCR技术.........................................................................................................12 系列二 Southern杂交技术........................................................................................14
实验一 植物总DNA的快速少量抽提(CTAB法)........................14 实验二 总DNA质量检测及酶切....................................................................15 实验三 电泳、转膜....................................................................................................16 实验四 Southern Blotting......................................................................................18 系列三 Northern Blotting............................................................................................24
实验一 RNA Extraction (mini prep).................................................................24 实验二 RT-PCR (Reverse transcription PCR)............................................26 实验三 RNA的电泳,转膜和杂交................................................................28 附录 试剂配方.....................................................................................................................29
一 细菌培养试剂..........................................................................................................29 2
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心二 质粒抽提试剂..........................................................................................................30
三 DNA操作试剂........................................................................................................31
四 RNA操作试剂........................................................................................................34
Stock Solution:...............................................................................................................34
Work Solution.................................................................................................................35

3
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心
系列一 分子克隆技术
实验一 质粒的制备
质粒是携带外源基因进入细菌中扩增或表达的主要载体,它在基因操作中具有重要作用。质粒的分离与提取是最常用、最基本的实验技术。
质粒的提取方法很多,大多包括3个主要步骤:细菌的培养、细菌的收集和裂解、质粒DNA的分离和纯化。本实验以碱裂解法为例,介绍质粒的抽提过程。
实验目的:掌握碱裂解法抽提质粒的原理、步骤及各试剂的作用。
实验材料:含有质粒pUC18载体的大肠杆菌菌液,克隆有水稻外源片段的BAC的大肠
杆菌菌液。
实验原理:在pH 12.0 ~ 12.6碱性环境中,细菌的线性大分子量染色体DNA变性分开,
而共价闭环的质粒DNA虽然变性但仍处于拓扑缠绕状态。将pH调至中性
并有高盐存在及低温的条件下,大部分染色体DNA、大分子量的RNA和蛋
白质在去污剂SDS的作用下形成沉淀,而质粒DNA仍然为可溶状态。通过
离心,可除去大部分细胞碎片、染色体DNA、RNA及蛋白质,质粒DNA
尚在上清中,然后用酚、氯仿抽提进一步纯化质粒DNA。
实验步骤:
1. 取含有pUC18质粒的大肠杆菌菌液于LA培养基上37℃过夜培养;
2. 用无菌牙签挑取单菌落,接种于含有Amp抗生素的LB培养基中,37℃摇床~250
r/min过夜培养;
3. 吸取1.5 ml菌液,12000 g离心2分钟,收集菌体,倒掉菌液;吸取1.5 ml菌液,
再次收集菌体,尽量将菌液倒干净;
4. 加入300 ml溶液 I振荡打匀,重新悬浮细胞,震荡混匀(注意:应彻底打匀沉淀或
碎块);
5. 加入300 ml溶液II,轻柔颠倒混匀,放置至清亮,一般不超过5分钟;
6. 加入300 ml溶液III颠倒混匀,放置于冰上10分钟,使杂质充分沉淀;
7. 12000 g离心10分钟;
8. 吸取800 ml上清液(注意:不要吸取到飘浮的杂质)至另一Eppendorf管中,加入
2/3体积的异丙醇,室温下放置5分钟;
9. 12000 g 常温离心15分钟;
4
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心10. 倒尽上清,加75%乙醇浸洗除盐(放置片刻或离心3分钟后倒去上清);
11. 室温放置或超净台上风干DNA;
12. 加40 ml灭菌超纯水或TE溶解;
13. 质粒、BAC的质量检测,于-20℃保存。
附注:质粒检测
电泳检测:质粒电泳一般有三条带,分别为质粒的超螺旋、开环、线型三种构型 吸光值检测:采用分光光度计检测260nm、280nm波长吸光值,若吸光值260nm/280nm的比值介于1.7-1.9之间,说明质粒质量较好,1.8为最佳,低于1.8说明有蛋白质污染,大于1.8说明有RNA污染。
实验二 DNA的琼脂糖凝胶电泳
带电荷的物质在电场中的趋向运动称为电泳。电泳的种类多,应用非常广泛,它已成为分子生物学技术中分离生物大分子的重要手段。琼脂糖凝胶电泳由于其操作简单、快速、灵敏等优点,已成为分离和鉴定核酸的常用方法。
实验目的:掌握琼脂糖凝胶电泳的原理,学习琼脂糖凝胶电泳的操作。
实验材料:质粒DNA、BAC、植物总DNA或它们的酶切产物。
实验原理:在pH值为8.0~8.3时,核酸分子碱基几乎不解离,磷酸全部解离,核酸分子
带负电,在电泳时向正极移动。采用适当浓度的凝胶介质作为电泳支持物,
在分子筛的作用下,使分子大小和构象不同的核酸分子泳动率出现较大的差
异,从而达到分离核酸片段检测其大小的目的。核酸分子中嵌入荧光染料(如
EB)后,在紫外灯下可观察到核酸片段所在的位置。
实验步骤:
1.用胶带将洗净、干燥的制胶板的两端封好,水平放置在工作台上;
2.调整好梳子的高度;
3.称取0.24 g琼脂糖于30 ml 0.5×TBE中,在微波炉中使琼脂糖颗粒完全溶解,冷却至45-50?C时倒入制胶板中;
4.凝胶凝固后,小心拔去梳子,撕下胶带;
5.将电泳样品与溴酚蓝混合后将样品依次点入加样孔中;
5
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心pUC18 5?l + ddH2O 3?l + 溴酚蓝2?l 共10?l于0.5ml tube中混合后点样;
6.将制胶板放入电泳槽中,加入电泳液,打开电泳仪,使核酸样品向正极泳动;
7.电泳完成后切断电源,取出凝胶,放入0.5 ?g/ml的溴化乙锭(EB)溶液中染色10-15 min,清水漂洗后置于紫外透射仪上观察电泳结果,并照相记录。
附注:
1.影响DNA在琼脂糖凝胶中迁移速率的因素:
1) DNA分子大小 迁移速率U与logN成反比(N为碱基对数目)。分子大小相等,电荷基本相等(DNA结构重复性)。分子越大,迁移越慢。等量的空间结构紧密的电泳快(超螺旋>线性DNA)
2) 琼脂糖浓度:logU=logU0 Krt 胶浓度,U为迁移率,U0 为DNA的自由电泳迁移率,t为胶浓度,Kr为介质阻滞系数。不同的凝胶浓度,分辨不同范围的DNA Agarose: 0.5%: 1-30 kb;
1.2%: 0.4-7 kb; 0.7%: 0.8-12 kb 1.5%: 0.2-3 kb.
3) DNA构象:一般迁移速率超螺旋环状>线状DNA>单链开环。当条件变化时,情况会相反,还与琼脂糖的浓度、电流强度、离子强度及EB含量有关。
4) 所加电压:低电压时,线状DNA片段的迁移速率与所加电压成正比。使分辨效果好,凝胶上所加电压不应超过5V/cm
5) 碱基组成与温度:一般影响不大4 -30 ℃
6) 嵌入染料的存在:降低线性DNA迁移率,(不提倡加在电泳液中)
7) 电泳缓冲液 (0.5×TBE)的组成及其离子强度影响DNA的迁移率,无离子存在时,核酸基本不泳动,离子强度过大产热厉害,熔化凝胶并导致DNA变性,一般采用1×TAE,1×TBE,1×TPE(均含EDTA pH8.0)。
2.溴化乙锭(EB)为致癌剂,操作时应戴手套,尽量减少台面污染。
3.电泳指示剂:核酸电泳常用的指示剂有两种,溴酚蓝(bromophenol blue, Bb)呈蓝
紫色;二甲苯晴(xylene cyanol, Xc)呈蓝色,它携带的电荷量比溴酚蓝少,在凝胶中的的迁移率比溴酚蓝慢。
实验三 外源DNA片段在质粒载体中的克隆
DNA重组技术包括载体及外源DNA片段的酶切消化、目的片段的获得及纯化、 6
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心目的片段与克隆载体的体外连接、重组子的筛选和鉴定等内容。DNA片段的克隆技术是分子操作的核心部分。
实验目的:学习DNA的酶切、纯化及外源片段与载体的连接,将BAC克隆所携带的外
源DNA酶切片段亚克隆到pUC18载体上。
实验材料:外源片段来自一个含有水稻DNA片段的BAC克隆的酶切片段;克隆载体为
PUC18。
实验原理:限制性内切酶可识别特定位点并切割DNA产生粘性末端或平端的外源片段,
经DNA的纯化处理后用于连接反应;选择克隆载体pUC18多克隆位点上
相应的限制性内切酶切割,并用碱性磷酸酶处理防止载体自连;在连接酶的
作用下将外源片段连接到载体上,实现外源片段的克隆。
实验步骤:
1.载体pUC18和外源DNA片段的限制性酶切:
(50 ml反应体系,用1.5ml tube,冰上操作):
DNA
R.E
30 ml 1 ml 5 ml 14 ml 10×buffer ddH2O
37℃反应1hr,分别取8 ml 外源片段酶切产物和5 ml PUC18酶切产物于1.0%凝胶检测酶切是否完全;按2-5步纯化、回收DNA
2.加入ddH2O 150 ml (扩大体积),加入等体积氯仿/异戊醇(24:1),颠倒混匀,12000g离心10 min;
3.吸取上清,加1/10体积3M NaAc和两倍体积无水乙醇,-20℃放置15分钟以上;
4.12000g 4℃冷冻离心15分钟;
5.倒去上清,用75%乙醇浸洗沉淀,风干后外源DNA溶于10 ml ddH2O(0.5ml tube中),PUC18溶于20 ml ddH2O(1.5ml tube中);
6.按以下反应去除载体PUC18的5’磷酸基团,50℃反应30 min 以上
DNA
20 ml 0.5 ml 4.0 ml 15.5 ml CIAP(TaKaRa) 10 ? buffer ddH2O
7.70℃水浴10 min, 使CIAP失活;
8.按2-5步纯化载体,溶于10 ml ddH2O;
7
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心9. 连接反应(15 ml体系):
DNA
10 ml 2.5 ml 1.5 ml 1 ml
16℃水浴过夜
10.
11.
附注:
1. 根据试验目的和外源片段的不同可选用不同的载体,采用不同粘性末端的双酶切可
实现外源片段的定向克隆。
2. 克隆中用到的几种工具酶:
(1)限制性内切酶
限制性内切酶的一个活性单位(1U):指在50 ml反应体系中,37℃下,经过1小时的反应将1mg DNA完全切割所需要的酶量。
限制性内切酶的star活性:限制酶在某些条件下使用时对DNA切割的位点特异性可能降低,即可以切割与原来识别的特定DNA序列不同的碱基序列,这种现象叫限制酶的star活性。它的出现与限制酶、底物DNA以及反应条件有关。
(2)碱性磷酸酶
细菌碱性磷酸酶(BAP)和牛小肠碱性磷酸酶(CIAP)都能催化水解DNA、RNA、dNTP和NTP上的5’-磷酸残基。比较而言,CIAP更常用,因其可在70℃ 10’内加热灭活或通过苯酚抽提而变性失活,同时CIAP的活性比BAP的高10-20倍。它主要用于:(1)克隆时去除载体的5’-P,以防载体自连;(2)在用 Kinase进行5’末端标记前,去除DNA的5’-P。
(3)连接酶
体外催化磷酸二酯键的形成可使用两种酶:大肠杆菌连接酶和T4噬菌体连接酶,但几乎在所有的克隆中T4噬菌体连接酶都是首选的酶,因其能在正常的反应条件下能有效的将平端连接起来。
3. 氯仿对眼睛、皮肤、粘膜及呼吸道都有刺激作用,它是致癌剂并可损伤肝脏和肾脏,
操作时需戴手套、安全镜并在通风橱中进行。苯酚是强腐蚀剂,能引起严重烧伤。操作时应戴手套、安全镜、穿防护服,并在通风橱中进行。
8 pUC18 5 ? buffer T4 ligase (3U/ml) 转化大肠杆菌感受态细胞 转化子的鉴定
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心实验四 感受态细胞的制备
体外连接的DNA重组分子导入合适的受体细胞才能大量增殖。为了提高受体菌摄取外源DNA的能力,提高转化效率以获得更多的转化子,人们摸索出了不同的方法处理细菌,使其处于感受态。目前主要采用电转化法和CaCl2法将外源DNA导入受体细胞中,并需要相应地制备电转化感受态细胞和CaCl2感受态细胞。
实验目的:学习感受态细胞的制备过程
实验材料:大肠杆菌菌株DH5a或DH10B
实验原理:电转化法是利用瞬间高压在细胞上打孔,因而需用冰冷的超纯水多次洗涤处于对数生长前期的细胞,以使细胞悬浮液中应含有尽量少的导电离子。转化效率为109~1010 转化子/?g DNA;对于热激法,是利用冰冷的CaCl2处理对数生长期的细胞,可以诱导其产生短暂的“感受态”,易于摄取外源DNA。转化效率为106 ~107转化子/?g DNA。
CaCl2感受态细胞的制备实验步骤
1.前夜接种受体菌(DH5a或DH10B),挑取单菌落于LB培养基中37℃摇床培养过夜(约16小时);
2.取1ml过夜培养物转接于100ml LB培养基中,在37℃摇床上剧烈振荡培养约2.5-3小时(250-300rpm);
3.将0.1M CaCl2溶液置于冰上预冷;以下步骤需在超净工作台和冰上操作
4.吸取1.5ml培养好的菌液至1.5ml离心管中,在冰上冷却10分钟;
5.4℃下3000 g冷冻离心5分钟;
6.弃去上清,加入100ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮,在冰放置20分钟;
7.4℃下3000 g冷冻离心5分钟;
8.弃去上清,加入100ml预冷0.1M CaCl2溶液,用移液枪轻轻上下吸动打匀,使细胞重新悬浮;
9.细胞悬浮液可立即用于转化实验或添加冷冻保护剂(15% - 20%甘油)后超低温冷冻贮存备用(-70℃)。
9
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心电转化法制备大肠杆菌感受态细胞的实验步骤
1. 前夜接种受体菌(DH5a或DH10B),挑取单菌落于LB培养基中37℃摇床培养过夜;
2. 取2ml过夜培养物转接于200ml LB培养基中,在37℃摇床上剧烈振荡培养至OD600=0.6(约2.5-3小时);
3. 将菌液迅速置于冰上。以下步骤务必在超净工作台和冰上操作
4. 吸取1.5ml培养好的菌液至1.5ml离心管中,在冰上冷却10分钟;
5. 4℃下3000g冷冻离心5分钟;
6. 弃去上清,加入1500ml冰冷的10%甘油,用移液枪轻轻上下吸动打匀,使细胞重新悬浮;
7. 4℃下3000g冷冻离心5分钟
8. 弃去上清,加入750ml冰冷的10%甘油,用移液枪轻轻上下吸动打匀,使细胞重新悬浮;
9. 4℃下3000g冷冻离心5分钟
10. 加入20ml冰冷10%的甘油,用移液器轻轻上下吸动打匀,使细胞重新悬浮;
11. 立即使用或迅速置于-70?C超低温保存。
附注:
影响感受态细胞转化效率的因素及实际操作过程中应注意的事项:
1) 细菌的生长状态:实验中应密切注视细菌的生长状态和密度,尽量使用对数生长
期的细胞(一般通过检测OD600来控制。DH5a菌株OD600为0.5时细胞密度是5×107/ml);
2) 所有操作均应在无菌条件和冰上进行;
3) 经CaCl2处理的细胞,在低温条件下,一定的时间内转化率随时间的推移而增加,
24小时达到最高,之后转化率再下降(这是由于总的活菌数随时间延长而减少造成的);
4) 化合物及无机离子的影响:在Ca2+的基础上联合其他二价金属离子(如Mn2+或
Co2+)、DMSO或还原剂等物质处理细菌,可使转化效率大大提高(100-1000倍);
5) 所使用的器皿必须干净。迹量的去污剂或其它化学物质的存在可能大大降低细菌
的转化效率;
6) 质粒的大小及构型的影响:用于转化的应主要是超螺旋的DNA;
7) 一定范围内,转化效率与外源DNA的浓度呈正比;
10
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心实验五 质粒的转化及转化子的鉴定
质粒的转化是指将质粒或以它为载体构建的重组子导入细菌的过程。将连接产物转化到感受态细胞中,实现重组克隆的增殖,便于后续分子操作。可以采用多种方法筛选和鉴定目的克隆。
实验目的:掌握热激法或电转化法转化大肠杆菌感受态细胞及转化子的鉴定方法。 实验材料:外源片段与载体的连接产物;大肠杆菌感受态细胞。
实验原理:(1)热激法:大肠杆菌在0℃ CaCl2低渗溶液中,菌细胞膨胀成球形,转化
混合物中的DNA形成抗DNase的羟基-钙磷酸复合物粘附于细胞表面,经
42℃短时间热冲击处理,促进细胞吸收DNA复合物,在丰富培养基上生长
数小时后,球状细胞复原并分裂增殖。在被转化的细胞中,重组子基因得到
表达,在选择性培养基平板上可挑选所需的转化子。
(2)电转化法:外加于细胞膜上的电场造成细胞膜的不稳定,形成电穿孔,不仅有利于离子和水进入细菌细胞,也有利于孔DNA等大分子进入。同时
DNA在电场中形成的极性对于它运输进细胞也是非常重要的。
热激法转化实验步骤:
1.制备选择性培养基平板:在融化的250ml LA培养基中250 ml Amp(100mg/ml),250 ml X-gal (20mg/ml),25 ml IPTG (200mg/ml),混匀后倒入灭菌培养皿中;
2.取出3管制备好的感受态细胞,放在冰上融化;
3.每100 ml感受态细胞加入约20ng质粒DNA,3管分别加连接产物、标准超螺旋质粒DNA(阳性对照)及不加入任何DNA(阴性对照),用移液器轻轻吸打均匀,在冰上放置30分钟;
4.热击:将离心管放置42℃水浴,热击90秒,注意:勿摇动离心管;
5.冰镇:快速将离心管转移至冰浴,放置1-2分钟;
6.复苏:每管加400 ml SOC培养基,在37℃摇床温和摇动温育45分钟,使细菌复苏;
7.布皿:取适当体积均匀涂布于含有IPTG、X-gal、抗生素(Amp)的LA平板;
8.培养:倒置培养皿,于37℃培养12-16小时 即可观察到蓝白相间的菌落(其中白色菌落为含有外源插入片段的转化子,蓝色菌落是载体自连的转化子)
11
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心质粒电转化大肠杆菌感受态细胞操作步骤
1.制备选择性培养基平板:在融化的250ml LA培养基中250 ml Amp(100mg/ml),250 ml X-gal (20mg/ml),25 ml IPTG (200mg/ml),混匀后倒入灭菌培养皿中;
2.取出制备好的感受态细胞,放在冰上融化;
3.每管感受态细胞加入1ml连接产物,用移液器轻轻吸打均匀,置冰上;
4.电转化仪选择1800V作为输出电压;
5.将要转化的混合物加入预冷的1 mm的电转化杯中,立即按下按纽电击;
6.立即加1ml SOC培养基到转化杯中重悬细胞;
7.将细胞转入合适的培养管中37?C培养1小时;
8.吸取合适体积的菌液涂布已倒好的选择培养基平板;
9.37?C培养过夜,观察结果。
附注:
1.利用氨苄青霉素抗性筛选转化子时,用转化细胞铺平板的密度要低(90mm平板上不得超过105个菌落),同时37℃培养不应超过20小时,具氨苄青霉素抗性的转化体可将b-内酰胺酶分泌到培养基中,迅速灭活菌落周围的抗生素,从而导致对氨苄青霉素敏感的卫星菌落的出现。
2.鉴定转化子中是否含有外源DNA片段常用的方法有:
1) a互补;
2) 杂交筛选;
3) 插入失活(一些老质粒如pBR322等);
4) 小量提取质粒酶切检测、PCR检测
实验六 PCR技术
聚合酶链式反应(polymerase chain reaction, PCR)是一种体外核酸扩增系统,是分子克隆技术中的常用技术之一。PCR具有反应快速、灵敏、操作简便等优点,已广泛应用于分子生物学的各个领域。
实验目的:掌握PCR原理,学习PCR操作过程
12
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心实验材料:转基因水稻叶片总DNA,外源基因的特异引物
实验原理:PCR是在模板DNA、引物和dNTPs的存在下依赖于DNA聚合酶的酶促反
应。PCR技术的特异性取决于引物和模板结合的特异性。反应分为变性、
退火、延伸三步,经过一定的循环,介于两个引物之间的特异DNA片段得
到大量扩增。
实验步骤:
1. 调整模板浓度至10 ng/ml;
2. 按下列体系配制反应混合液,混匀,加一滴矿物油,离心5秒
Template DNA
10? buffer
2 ml (20 ng) 2.0 ml 1.5 ml 0.2 ml 0.2 ml 2.0 ml 0.2 ml 20 ml MgCl2(25mM) Primer F (10?M) Primer R (10?M) dNTPs(2mM) Taq(5U/ml)
3.PCR反应循环条件设置: Add ddH2O to
95℃ 3' 1 cycle
94℃ 1' 55℃ 1' 72℃ 1'30" 35 cycles
72℃ 8' 1 cycle
4℃ forever
4.检测:加2 ml溴酚蓝,混匀,短暂离心,取15 ml反应产物点样电泳;
5.在1%的琼脂糖凝胶上点样电泳;EB染色,紫外观察。
附注:
1.引物设计应具有特异性,依靠引物设计软件进行引物设计;引物分装成多管,不宜反复冻融多次;
2.PCR反应的各种成份不能遗漏,操作应戴手套,冰上操作;
3.根据引物的Tm值和扩增片段长度以及PCR仪的特性来设定PCR循环条件;
4.注意分析电泳检测PCR产物时出现拖带或非特异性扩增带、无DNA带或DNA带很弱的可能原因。
13
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心
系列二 Southern杂交技术
Southern杂交,通过用限制性内切酶消化基因组或其它来源的DNA,经过琼脂糖凝胶电泳按大小分离酶切所得的片段,随后DNA在原位发生变性并从凝胶转移到一固相支持物上(一般是硝酸纤维素膜或尼龙膜)。DNA转移至固相支持物的过程中各DNA的相对位置保持不变,用一定方法(如放射性同位素)标记的DNA探针与固着在膜上的DNA 杂交,经X-光片自显影显现出与探针DNA互补的DNA电泳条带的位置。
实验一 植物总DNA的快速少量抽提(CTAB法)
DNA分子是分子生物学研究的基本材料,依不同的实验目的可采取不同的抽提方法获取数量和质量不等的DNA。
实验目的: 了解植物DNA抽提的主要方法,掌握CTAB法快速抽提水稻DNA。 实验材料及试剂:水稻叶片,1.5×CTAB,氯仿/异戊醇(24:1),95%乙醇或无水乙醇等 实验原理:植物DNA的抽提常采用两种方法:
(1)SDS法: 离子去污剂,过程长,纯度高;
(2)CTAB法:该方法简便、快速,DNA产量高(纯度稍次,适用于一般分子生物学操作)。 CTAB是一种非离子去污剂,植物材料在CTAB的处理下,结合65°C水浴使细胞裂解、蛋白质变性、DNA被释放出来。CTAB与核酸形成复合物,此复合物在高盐(>0.7mM)浓度下可溶,并稳定存在,但在低盐浓度(0.1-0.5mM NaCl)下CTAB-核酸复合物就因溶解度降低而沉淀,而大部分的蛋白质及多糖等仍溶解于溶液中。经离心弃上清后,CTAB-核酸复合物再用70-75%酒精浸泡可洗脱掉CTAB。再经过氯仿/异戊醇(24:1) 抽提去除蛋白质、多糖、色素等来纯化DNA,最后经异丙醇或乙醇等DNA沉淀剂将DNA沉淀分离出来。
实验步骤:
1. 采集适量幼嫩叶片,用液N2研成粉末,0.4 g装入1.5ml离心管中(-20℃预冷)。
2. 预热1.5×CTAB到95℃,加1ml到装有叶片粉末的离心管中,混匀(防止冻融)。 14
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心3. 立即置于65℃水浴30min,每5分钟,上下颠倒1次。
4. 12000g离心5分钟。
5. 吸取上清液约600?l,加入等体积(600?l)氯仿/异戊醇(24:1),上下颠倒数次,至
下层液相呈深绿色为止。
6. 12000g离心5分钟。
7. 取450?l上清于一新1.5ml离心管,加入1ml 95%乙醇和45?l 10M NH4AC),混匀,
室温放置10min。
8. 12000 g离心10min,去上清,用75% EtOH浸洗沉淀,自然干燥约30 min。
9. 加入50?l 1/10 TE或无菌水(含20?g/RNase),置于4℃过夜,待DNA溶解后,检
测DNA浓度及质量。
注意事项:
1.尽量取材幼嫩叶片,如太老,酚类物质多,必须用10 mM的β-ME处理
2.研钵预冻,粉末至加CTAB前不要融化
3.24:1的氯仿/异戊醇抽提时动作应轻柔,转移用的枪头最好是剪宽了的
4.所用试剂必需灭菌,手套
思考: (1)DNA降解的可能原因
(2)提高DNA产量的措施
实验二 总DNA质量检测及酶切
实验目的:了解掌握检测DNA 质量的方法以及DNA定量的方法;了解影响DNA在琼
脂糖凝胶中泳动速率的因素;训练DNA的琼脂糖凝胶电泳操作及DNA的
限制性内切酶操作。
实验原理:参见系列一中实验二;限制性内切酶的特点:见“基因操作原理”。
实验材料及试剂:水稻总DNA或BAC克隆DNA,琼脂糖,限制性内切酶DraI,EcoRI,
EcoRV,HindIII
实验步骤:
1.取10?l DNA于0.8% 凝胶检测;
2.将DNA调节浓度至300-400 ng/?l ;
2.仔细阅读将所用的任何一种酶产品说明书,熟悉反应条件及酶切的贮存浓度(10U-50U/?l)厂家配套试剂;
15
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心3.计算据反应条件所需要的各种试剂准确用量:(0.5 ml tube中)
DNA(3-5?g)
10?l 1.5?l 0.8?l(冰上) 2.7?l 10? buffer reaction Enzyme (15 U/?l) ddH2O
4.37℃ 混匀,短暂离心; 温浴1-2 hrs (纯DNA) 或10 hrs(粗制DNA);
5.加入上样缓冲液终止酶切反应,也可65℃加热10 min使酶变性失活;
6.电泳检测酶切效率:
每个样品取1/10量用琼脂糖电泳检测,制胶及点样方法同上。
结果分析
若水稻DNA呈现均匀连续分布的一片,则酶切效果好,否则需重做;
DNA被切烂:DNA降解,重新提DNA;
DNA切不动:杂质多(多糖,蛋白质,酚类,有机溶剂等),重新纯化;
若是BAC克隆DNA,酶切后应出现多条很清晰的不同大小DNA带。
思考:
什么是酶星活性?如何避免?
影响酶切效率的因素?
EB指示剂原理?
实验三 电泳、转膜
转膜是把DNA从琼脂糖凝胶中转移到固相支持物(一般是尼龙膜)上固定,是进行各种后续研究(如RFLP分析,阳性克隆的筛选验证等所有涉及分子杂交的研究)的前提。
实验目的:掌握Southern blotting的原理及操作步骤
实验原理:
1. 转膜的方式:
向上的毛细管转移
16
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心向下的毛细管转移
同时向两张膜转移
电转移
真空转移
2. 固相支持物的种类及选择:
硝酸纤维素膜:非共价结合,易脆,易丢失DNA,< 500 bp的DNA无效,转膜前的工作(从提高转膜效率,利于转膜后使用等)
尼龙膜(带正电荷的)高强度,不易破损,具有较大的DNA结合容量,它能够吸附变性DNA,核酸以共价结合方式不可逆的结合在尼龙膜上。尼龙膜两边均有同样吸附DNA功能,无论用哪边均可以经久耐用,可反复利用10次以上(10-20次),经毛细管(毛细吸附)作用,把DNA从凝胶上转到膜上。DNA转到膜上是复制胶上的带型,在80-100℃真空干燥2-4 hrs,即可固定DNA。
3. 转移缓冲液(transfer buffer)的选择:
带正电荷的尼龙膜:可用高盐离子强度(SSC),但不能充分发挥膜潜能;0.4N NaOH,共价结合DNA是最大优点。
硝酸纤维膜:高盐离子强度促进DNA与膜结合(20×SSC),低盐离子强度导致小片段DNA在转移过程中丢失,pH>9,DNA不能与膜结合。
4.转膜时间(duration of transfer)约12 hrs
取决于毛细管系统,DNA大小,胶厚度(<5 mm)及浓度(<1%)。
DNA分子量大小决定时间长短,部分去嘌呤减小DNA,碱转移2hrs大部分结合到膜。
DNA转移的效率较难判断,只有在转膜结束后,通过EB染胶,及分子杂交才可以鉴别效率高低,但已无补救措施,因此,每一步应严格操作。
实验材料及试剂:酶消化好的DNA样品,尼龙膜,0.2N HCl,0.4N NaOH等 实验步骤:
1. 0.8%琼脂糖电泳
制胶:注意琼脂糖的质量,胶的浓度,厚度(<5mm)及均一性。一般大电泳槽配制250ml 0.8%琼脂糖凝胶,采用42孔梳子(经济,高效)
制样,点样:DNA样品中指示剂量稍多
电泳:一般1-1.5V/cm的电压, 使DNA迁移到适当距离,一般指示剂约移动10-11cm (大电泳槽:40V×12-15hrs,小电泳槽30V×4-5 hrs)
2.转膜前的准备:
依胶大小每块凝胶准备两张比胶稍大的滤纸(11×12.5 cm),两张用作盐桥的滤纸, 17
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心一张与胶同样大小的尼龙膜(10×10.5cm),两个玻璃盘、两块有机玻璃板、一根玻璃棒,比尼龙膜稍大的一叠吸水纸等。
3.一玻璃盘中加入足量的0.4N NaOH,放上洗净的玻璃板,按图示搭制盐桥(操作示范)。
4.凝胶的预处理:
a) 从电泳槽中移出凝胶置于塑料板上,用切胶板把胶切成适当大小,切去右上角(最后一个样品的最前端)作为电泳方向记号
b) 把凝胶翻面,放入加有足量的0.2N HCl玻璃盘中,轻轻摇动10min,使指示剂变黄色为止(脱嘌呤)
c) 倒去HCl溶液,加蒸馏水漂洗凝胶
d) 倒去蒸馏水,加0.4N NaOH中和
e) 同时在盐桥滤纸上洒些0.4 NaOH,立即将胶放在盐桥上(忌气泡)
5.胶的四周用塑料片与胶紧紧相连,防止短路(吸水纸与盐桥相接)
6.在胶面上倒足够量0.4N NaOH,小心放置膜(预湿0.4N NaOH)使膜覆盖整块胶(要求一次成功,不能移动)
7.膜上放2张滤纸,滤纸大小为15×12cm
8.放不少于5cm厚的吸水纸,放上玻板,其上压约500g的重物,转膜12 hrs左右
9.转膜完毕,用2×SSC漂洗膜两次,各五分钟。用EB染胶以检测转移效果。
10.用两张滤纸包住膜,置于80-100℃的真空干燥箱中,干燥2-4 hrs。
思考:
(1)为什么转膜前要对凝胶预处理?如何处理?
(2)怎样提高转移效率
实验四 Southern Blotting
对于大的基因组,DNA酶切图谱凭肉眼是分辨不开的(EB染色),因为大小不等的分子呈现弥散分布,只有借助灵敏的放射性同位素(或其他化学发光物质),将靶DNA在凝胶上(膜上)的带型通过特定的探针与之杂交,转换成X光片上直观的带型,才能进行相关分析。另外如果需要鉴定或寻找与已知DNA同源的DNA片段如:染色体步查、基因组文库的评价和利用、阳性克隆的分析鉴定、转基因拷贝数分析等也都需进行DNA的分子杂交实验。
18
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心
实验目的:掌握同位素的操作及防护方法;掌握预杂交、探针的标记及分子杂交技术 实验原理:依据碱基配对原则,用放射性同位素标记的DNA探针,与固着在膜上的靶
DNA杂交,经放射自显影,确定靶DNA的位置。
1.预杂交:膜上有许多没有结合DNA分子的地方,若不在杂交前用一些封闭剂结合位
点,加入探针后,探针DNA分子将会结合在这些位点上,导致杂交背景深,
预杂交的目的是用非特异性DNA分子(鲑精DNA)及其它高分子化合物
(封闭剂)将待杂交膜中的非特异性位点封闭,从而减少杂交背景。
2.探针的标记:体外标记DNA或RNA的方法有多种,如:末端标记,随机引物标记,
缺刻平移(nick translation),体外转录(in vitro transcription)及各类PCR
等。这些方法有的是在特定位置标记核酸(5’或3’末端),有的标记核酸分
子内部的多个位点。有的产物是标记单链,有的产物是标记双链。有的方法
产生一定长度的标记产物,有的得到的是长短不一的标记产物。
随机引物标记:在DNA聚合酶的作用下,寡核苷酸通过与单链的模板配对可以启动DNA
的合成。如果寡核苷酸序列是不同的(heterogeneous),引物中包含所有可
能的随机序列(如6碱基引物则有46=4096种),可以与任意模板序列相配
对在许多位置形成杂交链,四种核苷酸底物中有一种是用同位素标记的,因
而可产生均匀一致高比活放射性探针。同位素标记的探针DNA 的平均长度
与引物的浓度成反比, The klenow fragment去除了E.Coli DNA polymerase
I 的5’→ 3’的外切酶活性,具有5’→ 3’的聚合酶活性及3’→ 5’ 外切酶活性;
Primer:可通过用DNase I消化牛胸腺DNA,DNA合成仪合成或直接从公司购买。同
位素标记的探针DNA的平均长度与引物的浓度成反比 [=k/(lnPc)1/2, Pc是
引物的浓度],一般可产生约400-600 bp的标记产物。
模板DNA:线状双连DNA。环状DNA用限制性内切酶切成线状,再标记。用纯化的
DNA片段作模板标记的探针与用完整的质粒作模板比较,可减少背景。
dNTPs: 其中一种用放射性同位素标记
3.杂交: 将标记好的探针,加到杂交液中,在一定(温度下)条件下,使探针与膜上
的DNA杂交。
4.洗膜:用不同组成及离子强度的(严谨度)溶液,洗去膜上的封闭剂及非特异性杂
交的探针
实验材料及试剂:已转移上DNA的尼龙膜,32P标记的dCTP,随机引物,20×SSC,
10%SDS,X-光片,显影液及定影液等
实验步骤:
19
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心1. 预杂交:将尼龙膜在2×SSC中浸湿,放入杂交袋或杂交管中,加入适量杂交液(杂
交液浸没膜),赶气泡,封口,放入65℃的杂交箱或恒温摇床中,预杂交3个小时以上,一般6-12hrs。
2. 标记探针:
反应体系:1-2blots(19.5×9.5cm2)
DNA
DNTP
100ng 2.0?l 5.0?l 1?l 1.0?l Random primer Klenow (1u/?l) a- dCTP*
add ddH2O to 17.0?l
按先将水和DNA 按反应体系所需的量取至一1.5毫升离心管中,混匀,短暂离心至管底,放至100℃干浴中变性10 min,变性探针迅速置于冰浴上5 min,按反应体系将反应混合液(dNTP,Random primer,Klenow )加入变性DNA中,在同位素操作台上,加入32P标记的dNTP(a-32P dCTP*,放射性比活>3000Ci/mmol),30℃温育3小时以上。
3. 杂交
将标记好的探针,补加300?l杂交液,100℃变性(or 0.4N NaOH变性),加入杂交袋(盒)中(忌直接加于膜上)。杂交前作标记效率测定,>25%以上可以往下做杂交, 过夜杂交。杂交效率受杂交速率及杂交稳定性影响。
4. 洗膜:从低严谨度到高严谨度冼膜液(具体情况而定):
1×SSC/0.1%SDS洗膜两次(冷5min,热65℃,15min)?检测信号强度
?0.5×SSC/0.1%SDS 热冼 65℃, 15min ?依情况可有改动0.2×SSC/0.1%SDS or 0.1×SSC/0.1%SDS。
5. 包膜:膜从洗膜液中捞出,在滤纸上晾干,膜表面无可见水膜为止(注意:不能太
干,以防探针难以洗脱影响再次使用),用保鲜膜包膜,压X-光片,置于-20或-70℃ 3—7天(依据信号强弱掌握曝光时间)
6. 冲洗X-光片
在暗室红灯下取出X-光片,置入显影液中至杂交带显现出来(显影时间依据信号强弱及曝光时间长短可由几秒钟到2分钟),转入清水中漂洗,然后放入定影液中定影至清亮(约10分钟)。自来水冲洗干净后,晾干,读片。
7. 膜上探针洗脱
再次使用膜前,必须洗去上次探针!
20
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心(1) 0.1%SDS,0.1×SSC
(2) 0.1NaOH,0.2%SDS 10min 2-3min
(3) 0.2M Tris.HCL, 0.2%SDS,0.1×SSC 20min
只洗去探针,而不影响膜上的靶DNA(因为DNA与膜是共价键结合,而DNA与探针的结合是氢键结合)。
附注:
1. 杂交效率的影响因素
a) 杂交温度:双链DNA分子,T=Tm-20—25℃可达最大杂交率,DNA-RNA杂交分子则低于Tm 值10-15℃。
b) 离子强度(1.5M/L NaCl杂交率最高)
c) 双链长度(形成杂交物长度) :杂交率与双链长度成正比
d) 探针的复杂程度 (重复性探针可以增加杂交率)
e) pH5.0-9.0基本无影响。
2. 影响杂交稳定性(影响解链温度的)因素
a) 离子强度 在0.01-0.4M NaCl之间, 每-10×单价阳离子,Tm-16.6℃
b) 碱基组成 AT<CG(在NaCl溶液中);
c) 去稳定剂(destabilizer) DNA-DNA杂交分子,每1%formamide,Tm?0.6℃;6M urea可降低Tm30℃
d) 碱基错配:每 1%的错配可使Tm降低1℃
e) 双链长度(探针杂交物) >500bp基本无影响
3.整个实验过程中应注意的事项:
1) 保证转膜质量;
2) 操作规范;
3) 提高杂交灵敏度(信号强度);
a) 探针量及标记量(探针变性);
b) 比活(性)度 >=108dpm/u(<108弱);
c) 靶DNA量(绝对量,酶切转膜决定;相对量,靶DNA相对于探针过量时,完全配对杂交,探针过剩时,完全配对和非严格的完全配对均有发生);
d) 有惰性聚合物增加灵敏度;10%(w/v)500,000(mw)dextran sulfate或8%(w/v)PEG6000。对于单链探针,可以增加10-fold杂交信号,dsDNA成
100-fold地增加杂交信号
4) 提高特异性:
a) 高盐溶液促进探针与靶序列的碱基配对20×SSC 3M NaCl/0.3M Na3Ci
21
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心b) 杂交后洗膜温度T?Tm
c) 杂交后洗膜液浓度组成,高严谨度洗膜液使不完全配对的杂交失去稳定,致使探针脱落
d) 杂交时间8hrs 以后,DNA探针逐渐退火,少量自由与靶DNA杂交.
e) 探针长度(>1000bp)过长,高严谨度下难洗脱非完全配对杂交探针。
4.放射性同位素:
1) 放射性同位素发出的射线主要有:
a粒子:外照射,一般能量的α粒子穿透能力较弱,射程短,危害性小,稍加防护
即可(如手套);内照射,电离密度大,危害大。
b粒子:穿透能力比α粒子强,外照射危害比α粒子大,可引起皮肤的放射损伤。 g射线:穿透能力很强,外照射时危害性很大,应采取切实有效的防护措施。
2) 放射性活度及单位:
放射性活度A是指一定量的放射性核素在时间间隔dt内发生自发核衰变数dN与此时间间隔的比值。即A=dN/dt
放射性活度的单位是Becquerel(贝克勒而),简称Bq。
1Bq=1个衰变/秒;1居里=3.7×1010Bq
其不表示放射出射线的多少(如60Co一个原子衰变防出1个b粒子和一个g光子,而一个32P原子只衰变出一个b粒子),也不表示射线能量的大小。
3) 辐射防护的目的:
防止发生对健康有害的确定性效应(接受放射性治疗的患者除外), 并将随机性损害效应的发生降低到被认为可以接受的水平,从而保障放射工作人员、公众及其后代的健康与安全,提高放射防护措施的效益,促进放射工作的发展。
4) 放射防护的原则:
a) 辐射实践的正当化:生产必须,医疗必须,科研教学必须
b) 辐射防护的最优化:即综合考虑社会、经济等诸因素之后,使个人剂量的大小、受照人数的多少和不确定发生的照射事件的发生概率可合理达到的低水平。
c) 个人剂量限制:为了保证每个人不致受到不合理的危害,必须制定一个个人剂量限制值,放射工作人员的剂量限制:全身均匀照射的年当量剂量限值H全身=50mSV; 不均匀照射时,有效剂量E不应超过H全身。
5) 外照射的防护措施:
a) 距离防护:人体受到照射的剂量率随着离开电离辐射源的距离的增大而减少。剂量率与距离的平方成反比。
b) 时间防护:在剂量率不变时,剂量与时间成正比,即操作时间越短,人员所受到的 22
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心照射剂量越小。( 要求放射性作业应操作熟练、操作步骤应尽量简单易行,尽量减少在辐射场所逗留的时间。)
c) 屏蔽防护:利用射线通过物质时,与物质相互作用使其能量被物质吸收而逐渐减弱的原理,可以设置一定的屏障物来进行防护。常用的材料有水、砖、大理石、混凝土、重金属铅等。
d) 利用衰变:可利用放射性物质存在自发衰变,其活性随之减少的原理进行外照射防护。如:半衰期小于15天的放射性废物,允许放置10个半衰期后作一般废物处理。
6) 内照射的防护措施
防止放射性物质经呼吸道吸入
防止放射性物质食入
防止放射性物质经体表进入
23
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心系列三 表达检测
在转录水平上研究和了解基因的表达与调控是分子生物学和基因操作的重要内容。Northern 杂交可以测定总RNA或poly(A)+ RNA 中特定mRNA分子的大小和表达丰度,RNA分子在变性琼脂糖凝胶中电泳,按其分子的大小分开,然后将RNA转至尼龙膜或硝酸纤维素膜上,经过体外铰链固定,用放射性同位素标记的DNA或RNA探针与之杂交,放射自显影后即可以得到待测基因RNA表达水平的情况;RT-PCR以mRNA 反转录生成的cDNA作为PCR的模板进行扩增,比Northern杂交更灵敏,对RNA的质量要求较低,操作简便,它是在转录水平上检测基因时空表达的常用方法。
实验一 RNA Extraction (mini prep)
RNA的制备与分析对于了解基因在转录水平上的表达与调控和cDNA的合成都是必须的,RNA的纯度和完整性对于Northern blot,RT-PCR和cDNA文库的构建等分子生物学实验都至关重要。RNA分离的方法很多,其中最关键的因素是尽量减少RNA酶的污染
方法一:氯化锂沉淀法
实验目的:学习RNA的简易制备过程,通过RNA电泳带评价RNA质量
实验材料:水稻幼嫩叶片
实验原理:SDS是一种去污剂,可以抑制内源RNA酶的活性,它不但可解聚核酸与蛋
白质的结合,还可与蛋白质带正电荷的侧链结合,形成SDS-蛋白质复合
物而沉淀。4 M LiCl可选择性沉淀RNA。
实验步骤:
1. Harvest fresh leaf discs in 2 ml eppendorf tubes and freeze quickly in liquid nitrogen and store at -80?C until use.
2. Grind leaf discs using a steel bar (precooled in liquid nitrogen) that perfectly fits the eppendorf tube. Keep the plant material frozen allows easy grinding to a fine powder.
3. After grinding , add 500ml of hot extraction buffer (80℃) and phenol (1:1 ) (250 ml of each). (Extraction buffer: 0.1 M LiCl, 100 mM Tris.HCl pH=8.0, 10 mM EDTA, 1% SDS)
4. Homogenized the mixtures by vortex for 30 seconds, and add 250 ml 24
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心chloroform/isoamylalcohol (24:1) and vortex.
5. Centrifuge for 5 min, remove the waterphases and mix with one volume of 4 M LiCl.
6. Store RNAs at 4℃ overnight and centrifuge for 10 min.
7. Dissolve the pellets in 250 ml DEPC water, add 0.1 vol. of 3 M NaOAc (pH 5.2), and precipitate the RNAs with 2.5 vols of ethanol.
8. Centrifuge for 10 min for 4℃, wash the RNA pellets with 70% EtOH and dry the pellets.
9. Dissolve the pellets in 15 ml DEPC water.
(Routinely between 25 and 50 mg of total RNA is obtained from 100 mg tobacco, tomato or potato leaf tissue)
方法二:Trizol法
实验原理:Trizol 试剂是由苯酚和硫氰酸胍配制而成的单相的快速抽提总RNA的试剂,在匀浆和裂解过程中,能在破碎细胞、降解细胞其它成分的同时保持RNA的完整性。在氯仿抽提、离心分离后,RNA处于水相中,将水相转管后用异丙醇沉淀RNA。 用这种方法得到的总RNA中蛋白质和DNA污染很少,可以用来做Northern,RT-PCR,分离mRNA,体外翻译和分子克隆等。
实验步骤:
1.液氮研磨样植物叶片,每1.5ml tube分装0.1克样品;
2.每管加1毫升Trizol 试剂(样品体积不超过Trizol 试剂体积的10%),迅速混匀(若样品较多可先将混好的样品置于冰上);室温下净置5-10分钟以利于核酸蛋白质复合体的解离;
3.加200 ?l的氯仿,用手剧烈摇荡15秒,室温下静置5分钟左右;
4.2-8 ℃ ,12000×g 离心10-15分钟;
5.将水相(上清)转入一新离心管,加0.5ml 异丙醇室温下沉淀10分钟;
6.2-8 ℃ ,12000×g 离心15分钟;
7.弃上清,加1ml 75%乙醇清洗RNA,振荡片刻后(务必使沉淀悬浮起来,以确保洗涤干净),7500×g 离心5分钟,小心弃上清;
8.室温静置5-15分,使RNA沉淀恰好干燥,加入20 ?l DEPC水溶解,取2 ?l 琼脂糖电泳检测RNA质量。采用分光光度计测定RNA浓度,将样品保存于-70 ℃超低温冰箱中备用。
注意事项:
25
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心抽提及操作RNA中要谨防RNase的污染,因此操作RNA的试剂及器皿都要进行相应的处理。
RNase-free water:Draw water to RNase-free glass bottles. Add diethylpyrocarbonate (DEPC)to 0.01% . Let stand overnight and autoclave.
研钵处理:0.4N NaOH 浸泡过夜,DEPC水洗涤3遍;
应戴口罩和手套,环境洁净;玻璃器皿180?C烘烤3hrs以上;塑料制品DEPC水清洗,蛋白质变性剂处理;一次性用品如tube,tip等用新的,高温高压消毒。
实验二 RT-PCR (Reverse transcription PCR)
实验目的:学习RT-PCR的原理及其操作过程。
实验材料:水稻叶片的RNA。
实验原理:目前PCR技术只能扩增DNA模板,对RNA模板不能直接扩增。mRNA 反
转录生成的cDNA可作为PCR的模板进行扩增,这种在mRNA反转录后进
行的PCR扩增称为RT-PCR。RT-PCR比Northern杂交更灵敏,对RNA的质量要求较低,操作简便,它是在转录水平上检测基因时空表达的常用方法。 本实验以水稻叶片RNA为材料,检测β-actin基因的表达。实验中设定2个
阴性对照:一个不加模板RNA,另一个不加反转录酶,主要是消除DNA及
PCR试剂方面引起的假阳性;同时以叶片DNA为阳性对照,检验PCR试剂
和扩增过程是否有问题。
实验步骤:
RNA Preparation
1. Prepare the plant total RNA by TRIzol Reagent.
2. Dilute the Total RNA to the final concentration of 1 mg / ml by DU640.
3. Combine the following in a 0.5 ml sterile eppendorf tube:
Total RNA
0.5-1mg 1ml (1u / ml)
1 ml
5 ml RNase-free DNase I 10× DNase I buffer DEPC-treated ddH2O
4. Incubate at 37℃ for 15 min, then 70℃ for 10 min.
Reverse Transcriptase Reaction
1. Add 1 ml 500 mg / ml oligo (dT) 15 primer, mix the contents of the tube by gently vortexing
and collect the reaction by brief centrifugation.
2. Heat the mixture at 65℃ dry bath for 10 minutes, then place at room temperature for 10
minutes. Add the following contents:
5 × first strand buffer
4 ml 26
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心0.1 M DTT
2 ml 1 ml
10 mM dNTP
3. Mix the contents of the tube by gently vortexing and collect the reaction by brief centrifugation. Place the tube at 37℃ water bath for 2 min.
4. Add 2 ml 200 U / ml M-MLV Reverse Transcriptase. Mix gently and incubate at 37℃ for 1 h. The total volume should now be 20 ml.
5. Inactive the reaction by heating at 70℃ for 15 min, then add 20 ml ddH2O and the first strand of cDNA can be used as a template to amplification in PCR.
6. To remove the RNA complementary to the cDNA, add 1 ml (2 units) of RNase H and incubate at 37℃ for 20 min. PCR Amplification
1. Prepare the mixture containing the following on ice:
cDNA first strand 10 × Ex Taq buffer Primer F (10 mM) Primer R (10 mM) ddH2O
2. Commence PCR program
94℃
Step 1 Step 2 Step 3 Step 4
3 min 1min
60℃
1 min
72℃
1 min 5 min
4℃
Cycles 1 30-40 1
1 ml 0.5 ml 5 ml 4 ml 2 ml 2 ml 35.5 ml
TaKaRa Ex Taq (5 u / 1 ml) dNTP mixture (2 mM)
forever
3. The β-actin is used as the internal standard for each RT-PCR, DNA contamination in the RNA sample is tested by replacing the reverse transcriptase with water in the RT-PCR. Reagents
5 × first strand buffer (GIBCO Part No.Y00146) 100 mM DTT (GIBCO Part No.Y11147)
200 U / ml M-MLV Reverse transcriptase (GIBCO Part No.28025-021) 500 mg / ml oligo (dT) 15 primer (Promega Cat. No. C110A 9362612) 10 mM dNTP mix
RNase-free DNase I (TaKaRa Code No. 2215 CA)
5 U / ml Ex Taq polymerase and 10 × buffer (TaKaRa Code No. RR001D CA) 10 ml M gene specific Primer F and R Sterile ddH2O Mineral oil
27
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心
实验三 RNA的电泳,转膜和杂交
实验原理:基本同Southern Blotting,但因RNA是单链分子,必须消除局部区域形成的
二级结构,在电场中泳动的距离才能反映它本身的大小,因而需使用变性胶
进行电泳;操作过程中应特别注意防止RNA的降解。
实验材料:经检测质量好的RNA样品
实验步骤:
1.制胶:
Agarose 1%-1.2%,1×MOPS buffer。
用灭菌的DEPC水定容,加热融化,冷却至70℃加甲醛,使终浓度为2%,通风橱中放1hr。
2.准备电泳液:1×MOPS buffer(小号槽约配500ml,中号槽约配900ml)
3.制样:测好浓度后按以下体系加样
15 mg RNA + 灭菌的DEPC水
Sample buffer
loading buffer
10 ml 8 ml 2 ml 0.03 ml EB (10mg/ml,专用于RNA)
65℃水浴变性10 min,立即放冰上,直至点样
4.点样:点样要仔细,不要漏出,并记录点样顺序。
5.电泳:中号槽: 电压100V电泳1hr左右,至溴酚兰离点样孔约3.5-4cm(凝胶在紫外灯下可看到28S和18S刚好分开即可)
6.转膜:转膜液用500ml 4×SSC加27ml 37%甲醛(终浓度为2%);转膜步骤同Southern。注意加溶液后一切操作均在通风橱内进行,以避免甲醛对人体的伤害
7.照相:将胶及膜取下,分别在凝胶成像系统下看胶上RNA是否有残留,膜上RNA效果是否完好。
8.风干:在超净工作台上吹干。
9.固定:于紫外交联仪内以1200 ENERGY照一次约半分钟,再重复一次,再在超净工作台上吹干。
10.
11.
杂交:在杂交管内加约50ml杂交液,将膜卷起放入,注意RNA面朝内,于64℃预杂交1-4hr 探针准备:预先挖胶回收的PCR产物或酶切产物(better),测浓度,跑胶验证28
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心后,按以下体系加入
DNA
3 ml(约200ng) 10 ml 4 ml 10 ml 2 ml 5 ml ddH2O DNTP(1.67mM) 杂交Buffer α-dCTP* Klenow Fragment
先加入DNA 和 ddH2O,100℃变性10min,冰上放5min,再加入DNTP和杂交 primer,加酶时注意低温操作,先加在管壁上,立即到同位素室加同位素,混匀离心,于室温下反应半小时。
12. 探针纯化:在原反应体系中继续加入
ddH2O 66 ml
NaAc 10 ml
无水乙醇 250 ml
混匀离心12000rpm 5min,倒去上清后加500 ml无水乙醇洗,离心 2 min,12000 rpm,倒去上清,检测信号,达103-104较好,加40 ml ddH2O和400 ml杂交液之后混匀离心,于100℃变性10min,冰上放置5min
13.
14. 13探针:取出杂交管倒去杂交液,加入15ml杂交液,将探针加入,盖紧,于64℃洗膜,压片:将杂交管中液体倒出,先加少量洗膜液Ⅰ,冷洗5min,倒出,再杂交过夜 多加些洗膜液Ⅰ,冷洗10min,将膜取出测信号,根据信号大小决定是否热洗,若信号高,用洗膜液Ⅱ,Ⅲ继续洗,直到信号值达到适宜值为止,即膜上某一区域信号强,而其他区域信号弱代表杂交上了,然后包膜压片,放于-20℃或-80℃3天左右即可洗X片。
15. 结果观察与分析
附录 试剂配方
一 细菌培养试剂
LB培养基
NaCl
Yeast extract
Peptone
10 g 5 g 10 g 29
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心Add dH2O to 1000 ml
Aliquot to 500ml flasks (250 ml per flask). Seal with sealfilm
and autoclave them. Store at room temperature.
LA固体培养基
NaCl 10 g
Yeast extract 5 g
Peptone 10 g
Agar powder 13~15 g
Add dH2O to 1000 ml
Aliquot to 500 ml flasks (250 ml per flask). Seal with sealfilm
and autoclave them. Store at room temperature.
X-gal (20mg/ml):
20mg X-gal溶于1ml 二甲基甲酰胺中,-20℃避光保存;
IPTG (200mg/ml):
1g IPTG溶于4ml去离子双蒸水中,定容至5ml,过滤灭菌后-20℃保存;
Amp(100mg/ml):
1g Amp溶于4ml去离子灭菌双蒸水中,定容至5ml,-20℃保存
二 质粒抽提试剂
Solution Ι
Cell resuspension solution (50 mM Tris-HCl, pH 7.5, 10 mM EDTA, RNase A 100 μg/ml)
1 M Tris-HCl (pH 7.6) 2.5 ml
0.25 M EDTA 2.0 ml
ddH2O 45 ml
sterile by autoclave
add 1% RNase 0.5ml
store at room temperature
Solution Ⅱ
Cell lysis solution (0.2 N NaOH, 1% SDS)
Mix 0.4 N NaOH and 2% SDS in same volume.
Solution Ⅲ
Neutralization solution (1.32 M potassium acetate, pH 5.2)
5 M potassium acetate 13.2 ml
Hac 27 ml
adjust pH to 5.2
add ddH2O to 50 ml
30
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心sterile by autoclave
store at room temperature
RNaseA: 10mg/ml溶于TE中,在沸水中煮10-30分钟之后分成小份储存于-20℃
三 DNA操作试剂
1.5×CTAB
CTAB 15 g
1 M Tris·Cl (pH 8.0) 75 ml
0.5 M EDTA 30 ml
NaCl 61.4 g
add ddH2O to 1000 ml
0.5 M EDTA (pH 8.0)
EDTA-Na·2H2O 186.1 g
NaOH ~20 g
Adjust to pH 8.0
dH2O to 1000 ml
sterilize by autoclaving
1 M Tris·HCl
pH 7.4 pH 7.6 pH 8.0
Tris base 121.1 g 121.1 g 121.1 g
Concentracted HCl ~70 ml ~64 ml ~42 ml
dH2O to 1000 ml 1000 ml 1000 ml
Sterilize by autoclaving
TE (pH 8.0)
Stock vol.
10 mM Tris·HCl (pH 8.0) 1 M 10 ml
1 mM EDTA (pH 8.0) 0.5 M 2 ml
dH2O to 1000 ml
sterilize by autoclaving
10 M NH4Ac
NH4Ac 385 g 770 g
H2O to 500 ml 1000 ml
10×PCR buffer
stock vol.
500 mM KCl 2.5 M(sterilized) 200 ml
31
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心100 mM Tris-HCl 1 M pH 9.0(sterilized) 100 ml
1% Triton X-100 100% 10 ml
ddH2O 690 ml
sterilize by autoclaving
5×TBE
Tris 54 g
Boric acid 27.5 g
0.5 M EDTA (pH 7.9) 20 ml
dH2O to 1000 ml
10×TAE
Tris 121.1 g 484.4 g
EDTA(0.5 M) 20 ml 80 ml
NaAc·3H2O 17 g 68 g
glacial acetic acid ~30 ml ~200 ml
adjust to pH 8.1
dH2O to 1000ml 4000ml
NaOH
10 N 4 N
NaOH 400 g 160 g
dH2O to 1000 ml 1000 ml
2 N HCl
concentrated HCl 365 ml 182.5 ml
dH2O to 2000 ml 1000 ml
5 mg/ml ssDNA
Salmon sperm DNA 1 g
ddH2O to 200 ml
0.5 M P.B (phosphate Buffer) pH 6.8
Na2HPO4 16.44 g 131.52 g
NaH2PO4 16.11 g 128.88 g
dH2O to 500 ml 4000 ml
20×SSC
NaCl 175.3 g 701.2 g
Na3Citrate 88.2 g 352.8 g
dH2O to 1000 ml 4000 ml
Sterilize by autoclaving
10% SDS
32
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心SDS 100 g
dH2O to 1000 ml
Heat to 68 ℃ to assist dissolution
50×Denhart’s Solution
Ficoll 400 10 g
PVP-360 10 g
BSA (Fraction V) 10 g
ddH2O to 1000 ml
Southern Blot Hybridization Buffer (Saghai,s Lab)
Final conc. Stock Vol.
5×SSC 20× 250 ml
50 mM PB (pH 6.8) 0.5 M 100 ml
5×Denhardt’s 50× 100 ml
2.5 mM EDTA (pH 8.0) 0.5 M 5 ml
100 μg/ml ssDNA 5 mg/ml 20 ml
0.4%SDS 20% 20 ml
Dextran sulfate 50 g
ddH2O to 1000 ml
(Place a beaker on a stirrer, add these solution in the order of appearance one by one. SDS should be the very last item.)
Washing off Probe for Re-hybridization of Blots (I)
Washing time: 10 min
Final conc. Stock Vol.
0.1×SSC 20×SSC 20 ml
0.1% SDS 10% SDS 40 ml
dH2O to 4000 ml
Washing off Probe for Re-hybridization of Blots (II)
Washing time: 3 min
Final conc. Stock Vol.
0.1 N NaOH 10 N NaOH 40 ml
0.2% SDS 10% SDS 80 ml
dH2O to 4000 ml
Washing off Probe for Re-hybridization of Blots(Ⅲ)
Washing time: 20 min
Final conc.
Stock Vol. 800 ml 20 ml 80ml 4000ml 0.2 M Tris. (pH 7.5) 0.1×SSC 0.2% SDS dH2O to
1 M Tris. (pH 7.5) 20×SSC 10% SDS 33
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心
Blue Juice
Final conc. Stock
70% Glycerol 100%
0.5×TBE 5×
0.2% SDS 10%
20 mM EDTA 0.5 M
5 mg/ml Bromphenol Blue
5 mg/ml Xylene cyanol
dH2O to Vol. 35 ml 5 ml 1 ml 2 ml 0.25 g 0.25 g 50 ml Vol. 70 ml 10 ml 2 ml 4 ml 0.5 g 0.5 g 100 ml
EB (10 mg/ml)
ehidium bromide 1 g
dH2O to 100 ml
Stir on a magnetic stirrer for several hours. Transfer the solution to
a dark bottle and store at 4℃.
The concentration of work solution: 0.5 μg/μl (50 μl stock solution
In 1000 ml dH2O).
Decontamination of EB
Reduce the concentration of EB <0.5 mg/ml, add 1 volume of
0.5 M KMnO4,mix carefully then add 1 volume of 2.5 N HCl,
mix carefully and allow the solution to stand at room temperature
for several hours. Add 1 volume of 2.5 N NaOH, mix and discard.
四 RNA操作试剂
Stock Solution:
1M NaAc(PH7.0):82g NaAc 先加一定量ddH2O,用NaOH调PH值至7.0,再用ddH2O定容至1L,灭菌
0.5M EDTA(PH8.0): 186.1g EDTA先加一定量ddH2O, 用NaOH调PH值至8.0,再用ddH2O定容至1L,灭菌
10×MOPS buffer: (用DEPC水配,再灭菌)
MOPS 41.85g
1M NaAc(PH7.0) 50ml
34
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心0.5M EDTA(PH8.0) 20ml
先加一定量DEPC水,再用4N NaOH 调PH 至7.0(约加7ml),再用DEPC水定容至1L,灭菌
1M NaH2PO4 buffer ( PH7.2):
NaH2PO4 71g
H3PO4(85%) 4ml
用灭菌的DEPC水定容至1L
20×SSC:
NaCl 175.3g
Na3Citrate 88.2g
用ddH2O定容至1L,灭菌
10%SDS:
SDS 100.0g
用灭菌的ddH2O定容至1L(将水加热到68℃有助于溶解)
Work Solution
Sample buffer:
Deionized formamide 1000 ml
10×MOPS buffer 200 ml
37% formaldehyde 320 ml
Blue juice(loading buffer):
Glycerol 70ml
5×TBE 10ml
10%SDS 2ml
0.5M EDTA(PH8.0) 4ml
Bromphenol Blue 0.5g
Xylene Cyanol 0.5g
加灭菌的ddH2O定容至100ml
Hybridization buffer:
1M NaH2PO4 buffer 500ml
0.5M EDTA(PH8.0) 2ml
10% SDS 70g
BSA 10g
用灭菌的DEPC水定容至1L,贮存于室温下
35
生物秀论坛——学术交流、资源共享、互助社区 /bbs
生物秀实验频道——生命科学实验中心洗膜液Ⅰ:(for 1 littre)
20×SSC 100ml
10%SDS 10ml
洗膜液Ⅱ:(for 1 littre)
20×SSC 25ml
10%SDS 10ml
洗膜液Ⅲ:(for 1 littre)
20×SSC 5ml
10%SDS 10ml
4×SSC:
20×SSC 200ml
用灭菌的DEPC水定容至1L
2×SSC:
20×SSC 100ml
用灭菌的ddH2O定容至1L
36
生物秀论坛——学术交流、资源共享、互助社区 /bbs