一、实习时间:2014.11.24----2014.12.05
二、实习地点:邯郸市食品质量技术监督检验所
三、实习目的:学生在学校学习和教学实习的基础上,为进一步从专业工作实际出发,学生到其就业意向单位进行生产实习。学生的生产实习原则上按照企业的实习要求进行顶岗生产实践训练,学校提供生产实习大纲,供企业参考使用,通过生产实习,使学生加深对企业的了解,培养对企业的感情,提高社会适应性,熟练岗位操作能力,帮助学生实现“零距离”上岗。
四、实习内容 :
(一)食品中环己基氨基磺酸钠(甜蜜素)的检定
(二)食品中山梨酸钾、苯甲酸、糖精钠的测定
(三)植物油中抗氧化剂丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)的测定(高效液相色谱法)
(四)花椒里罗丹明的测定
(五)测定植物油中的溶剂残留
(六)测水中的三氯甲烷、四氯化碳
(七)测定酒中的塑化剂(领苯二甲酸酯类)
(八)测定酒中的己酸乙酯
(一)食品中环己基氨基磺酸钠(甜蜜素)的检定
1 主题内容与适用范围
本标准规定了食品中环己基氨基磺酸钠的三种测定方法——气相色谱法、比色法、薄
层层析法。
本标准气相色谱法及比色法适用于饮料、凉果等食品中环己基氨基磺酸钠的测定;薄
层层析法适用于饮料、果汁、果酱、糕点中环己基氨基磺酸钠的含量测定,样品中含有
0.1g/kg环已基氨基磺酸钠即可检出,最低检出量为3~4μg环己基氨基磺酸钠。
气相色谱法
2 原理
在硫酸介质中环己基氨基磺酸钠与亚硝酸反应,生成环己醇亚硝酸酯, 利用气相色谱法进行定性和定量。
3 试剂
本标准所用试剂,凡未指明规格者,均为分析纯(AR),水为蒸馏水。
3.1 正己烷。
3.2 氯化钠。
3.3 层析硅胶(或海砂)。
3.4 50g/L亚硝酸钠溶液。
3.5 100g/L硫酸溶液。
3.5 环己基氨基磺酸钠标准溶液(含环己基氨基磺酸钠>98%):精确称取1.0000g环己
基氨基磺酸钠,加水溶解并定容至100mL,此溶液每毫升含环己基氨基磺酸钠10mg。
4 仪器
4.1 气相色谱仪附氢火焰离子化检测器。
4.2 旋涡混合器。
4.3 离心机。4.4 10μL微量注射器。
4.5 色谱条件
4.5.1 色谱柱:长2m,内径3mm,U形不锈钢柱。
4.5.2 固定相:Chromosorb W AW DMCS80~100目,涂以10%SE-30。
4.5.3 测定条件
柱温:80℃;汽化温度:150℃;检测温度:150℃。
流速:氮气40 mL/min;氢气30 mL/min;空气300 mL/min。
5 样品处理
5.1 液体样品:摇匀后直接称取。含二氧化碳的样品先加热除去,含酒精的样品加40g/L
氢氧化钠溶液调至碱性,于沸水浴中加热除去,制成试样。
5.2 固体样品:凉果、蜜饯类样品将其剪碎制成试样。
6 分析步骤
6.1 试料制备
6.1.1 液体试样:称取20.0g试样于100 mL带塞比色管,置冰浴中。
6.1.2 固体试样:称取2.0g已剪碎的试样于研钵中,加少许层析硅胶(或海砂)研磨至
呈干粉状,经漏斗倒入100 mL容量瓶中,加水冲洗研钵,并将洗液一并转移压容量瓶中,
加水至刻度,不时摇动,1h后过滤,即得试料,准确吸取20 mL于100 mL带塞比色管,置
冰浴中。
6.2 测定
6.2.1 标准曲线的制备,准确吸取1.00 mL环己基氨基磺酸钠标准溶液于100 mL带塞比色
管中,加水20 mL,置冰浴中,加入5mL 50g/L亚硝酸钠溶液,5mL 100g/L硫酸溶液,摇匀,
在冰浴中放置30 min,并经常摇动,然后准确加入10 mL正己烷,5g氯化钠,摇匀后置旋
涡混合器上振动1min(或振摇80次),待静止分层后吸出己烷层于10 mL带塞离心管中进
行离心分离,每毫升已烷提取液相当1mg环己基氨基磺酸钠,将标准提取液进样1~5μL于气相色谱仪中,根据响应值绘制标准曲线。
6.2.2 样品管按6.2.1自“加入5mL 50g/L亚硝酸钠溶液……”起依法操作,然后将试料
同样进样1~5μL,测得响应值,从标准线图中查出相应含量。
7 计算
m1×10×1000 10m1
X = ———————— = ————— ..................(1)
m×V×1000 m×V
式中:X——样品中环己基氨基磺酸钠的含量,g/kg;
m——样品质量, g;
V——进样体积,uL;
10——正己烷加入量,mL;
m1——测定用试料中环己基氨基硝酸钠的含量,μg。
8 精密度和允许误差
重复测定变异系数<7%。
两次平行测定相对允许误差绝对值≤10%,平行测定结果用算术平均值表示,保留二
位小数。
(二)食品中山梨酸钾、苯甲酸、糖精钠的测定
1 范围
本标准规定了食品中苯甲酸、山梨酸和糖精钠含量的高效液相色谱测定方法
本标准适用于食品中苯甲酸、山梨酸和糖精钠含量的测定。
本标准的检出限:对于固态食品,苯甲酸、山梨酸、糖精钠的检出限分别为1.8mg/kg、1.2mg/kg、3.0mg/kg
2 原理
不同样品经提取后,将提取液过滤,经反相高效液相色谱分离测定,根据保留时间定性,外标峰面积定量
3 试剂和材料
方法中所用试剂,除另有规定外,均为分析纯,水为重蒸水。
3.1 甲醇 色谱纯
3.2 乙酸铵溶液 称取1.54g乙酸铵,加水溶解并定容至1000mL,溶液经滤膜(0.45μm)过滤。
3.3 亚铁氰化钾溶液: 称取106g亚铁氰化钾,用水溶解,并稀释至1000 mL。
3.4 乙酸锌溶液:称取220g乙酸锌溶于少量水中,加入30ml冰醋酸,加水稀释至1000 mL
3.5 氨水(1+1)氨水加水等体积混匀。
3.6 正己烷
3.7 ph4.4乙酸盐缓冲溶液:
a)乙酸钠溶液:称取6.80g乙酸钠,用水溶解后定容至1000ml
b)乙酸溶液:取4.3ml冰乙酸,用水稀释至1000ml
将上述两种溶液按体积比37:63混合,即得ph4.4乙酸盐缓冲溶液
3.8: ph7.2磷酸盐缓冲溶液:
a)称取23.88g磷酸氢二钠,用水溶解后定容至1000ml
b)称取9.07g磷酸二氢钾,用水溶解后定容至1000ml
3.9: 标准溶液的配制
a) 苯甲酸标准溶液 准确称取0.2360g苯甲酸钠,加水溶解并定容至200mL。此溶液每毫升相当于含苯甲酸1.00mg
b) 山梨酸钾标准储备溶液 准确称取0.2680g山梨酸钾,加水溶解并定容至200mL。此溶液每毫升相当于含山梨酸1.00mg
c) 糖精钠标准储备溶液 准确称取0.1702g糖精钠,(120度烘干4h) 加水溶解并定容至200mL。此溶液中糖精钠的含量为1.00mg/ml
d)混合标准使用液 取不同体积苯甲酸、山梨酸、糖精钠标准储备溶液,将其稀释成苯甲酸、山梨酸、糖精钠的含量分别为0.000mg/mL、0.020mg/mL、0.040mg/mL、0.080mg/mL、0.160mg/m、L0.320mg/mL的混合标准使用液
3.10 微孔滤膜(0.45μm)水相
4 仪器与设备
4.1 高效液相色谱仪:配有紫外检测器.
4.2 离心机:转速不低于4 000 r/min.
4.3 超声波水浴振荡器。
4.4 食品粉碎机.
4.5 旋涡混合器
4.6 pH计.
4.7 天平:分度值为0.01 g和0. 1 mg.
5 分析步骤
5.1 样品处理
5.1.1液体样品
5.1.1.1碳酸饮料、果酒,葡萄酒等液体样品:称取10 g样品(精确0.001g)(如含有乙醇需水浴加热除去乙醇后再用水定容至原体积)于25 mL容量瓶中,用氨水 (1+1)(3.5)调节pH至近中性。用水定容至刻度。混匀。经微孔滤膜(3.10)过滤,滤液待上机分析。
5.1.1.2乳饮料、植物蛋白饮料等含蛋白质较多的样品:称取:10g样品(精确至0.00lg)于25 mL
容量瓶中,加人2 mL亚铁氰化钾溶液(3.3),摇匀,再加人2 mL乙酸锌溶液(4。4)摇匀,以沉淀蛋白质,加水定容至刻度。4 OO0r/min离心10 min。取上清液。经微孔滤膜过滤,滤液待上机分析。
5.1.2半固体样品
5.1.2.1 含有胶基的果冻样品:称取0.59一1g样品(精确至0.001g),加水适量,转移至25 mL容量瓶中,再加水至约20 mL,置60℃~70℃水浴中加热片刻,加塞,剧烈振摇使其分散均匀后,加氨水(l+1)(3 .5)调节pH至近中性,加塞.剧烈振摇,使样品在水中分散均匀,置60℃至70℃水浴锅中加热30分,取出后趁热超声5分,冷却后用水定容至刻度。用微孔滤膜(3.10)过滤.滤液待上机分析。
5.1.2.2油脂、奶油类样品:称取2g一3g样品(精确至0.001g) 于50 mL具塞离心管中,加人10 ml正己烷(3.6).用旋涡混合器使其充分溶解,4 000 r/min离心3in。吸出正己烷提取液转移至250 mL分液漏斗中。再向50ml具塞离心管中加人10mL正己烷重复上述步辍,合并正己烷提取液于250 mL分液漏斗中。于分液漏斗中加人20 ml,pH4.4乙酸盐缓冲溶液(4,7)加塞后剧烈振摇分液漏斗约30s静置分层后,将水层转移至50mL容量瓶中·再加人20mL pH4.4乙酸盐缓冲溶液,重复上述步骤,合并水层并用乙酸盐缓冲济液定容至刻度,经微孔滤膜(3.1.0)过滤,滤液待上机分析。
5.1.3固体样品
5 .1 .3.肉制品、饼干、糕点:称取粉碎均匀样品2g~3g(精确至0.001g)于小烧杯中,用20 mL水分数次清洗小烧杯将样品移人25 mL容量瓶中,超声振荡提取5分钟,取出后加2 ml亚铁氰化钾溶液(3.3),摇匀。再加人2 mL乙酸锌溶液(3 .4).摇匀.用水定容至刻度.移人离心管中,准4000r/min离心5 min,吸出上清液,,用微孔滤膜(3.10)过滤,滤液待上机分析。
5.1.3.2油脂含量离的火锅底料、调料等样品:称取样品2g~3g(精确至0.001g)于50 mL具塞离心管中。加人10 mL磷酸盐缓冲液(3 .8),用旋涡混合器充分混合,然后于400Or/min离心5
Min,小心吸出水层转移到25 mL容量瓶中,再加人10 mL磷酸盐缓冲液于具塞离心管中,重复上述步骤,合并两次水层液.用磷酸盐缓冲液定容至刻度,混匀,用微孔滤膜(3.10) ,过滤,滤液待上机分析。
5 .1 .3.3凝胶糖果、胶基糖果:按半固体样品6.1.2,1含有胶基的果冻样品处理。
5.2色谱条件
a) 色进柱:C18柱,250 rnm×4.6 mm,5um,或性能相当者,
b) 流动相:甲醇 (4.1)十乙酸铵溶液(4.2)(5+95)。
c) 流速:1 mL/min;
d) 检测波长:230 nm;
e) 进样量:10u L。
5.3测定
取处理液和混合标准使用液各10uL注人高效液相色谱仪进行分离,以其标准溶液峰的保留时间为依据定性.以共峰面积求出样液中被测物质含量.供计算(笨甲酸、山梨酸和糖精钠同事测定的谱图参考见附录A)。
6 结果计算
样品中苯甲酸、山梨酸和糖精钠的含量按式(1)计算:
X=c×V×1000/(m×1000)
式中:
X—样品种待测组分含量.单位为克每千克
c一由标准曲线得出的样液中待测物的浓度.单位为毫克梅毫升
V—样品定容体积.单位为毫升 ‘
m—样品质量,单位为克(g)。
计算结果保留两位有效数字。
在重复性条件下获得的两次独立测定的结果的绝对差值不得超过算术平均值的10%
(三)植物油中抗氧化剂丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)的测定(高效液相色谱法)
1 范围
本标准规定了高效液相色谱法测定植物油中丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)的含量的方法
本标准适用于植物油中丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)含量的测定
本方法的检出限:TBHQ为1.0mg/kg、BHA为1.0mg/k、gBHT为0.5mg/kg
2 规范性引用文件
下列文件中的条款通过本标准的引用而成为本标准的条软。凡是注日期的引用文件,其随后所有的修改单(不包括勘误的内容〕或修订版均不适用于本标准,然而,鼓励根据本标准达成协议的各方研究是否可使用这些文件的最新版本.几是不注目期的引用文件,其最新版本适用于本标准。
3 原理
样品中丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)经甲醇提取纯化,反相C18柱分离后,用紫外荧光检测器280nm检测,外标法定量
4 试剂
除非另有说明.均使用分析纯试剂和二级水。
4.1 甲醇:色谱纯
4.2 乙酸:色谱纯
4.3 混合标样储备液取丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ) 各100rnl用甲醉溶解并定量至100ml。
4.4 流动相.流动相A为甲醇,流动相B为1%乙酸水溶液。
5 仪器和设备
5.1 分析天平,感量0.0001g
5.2 离心机.转速为3000r/min
5.3 漩涡混合器。
5.4 15ml具塞离心管.
5.5 0.45um有机相滤膜.
5.6 Nova-pakC18色谱柱(3.9mm×150mm).
5.7 高效液相色谱仪,带紫外检测器。
6 分析步骤
6.1 试样制备
按GB/T15687制备样品。
6.2 试液的制备
准确称取植物油样的5g以精确至0.001g置于 15 mL具塞离心管中.加入8 mL甲醇(4.1),旋涡混合3min,放置2 min.以3000r/min离心5min.取出上清液于25mL容量瓶中.残余物每次用8mL甲醇(4.1)提取2次.清液合并于25mL容量瓶中.用甲醇(4.1)定容.摇匀,经0.45um有机相滤膜过滤,滤液待液相色谱分析.
6.3 色谱分析
取lmg/ml,混合标样储备液(4,3),用甲醇(4·1)稀释至10ug/mL、50ug/mL、100ug/rnL、150ug/ml、200ug/ml、250ug/ml标准使用液连同样品依次进样,进行液相色谱检测,建立工作曲线。
色谱条件:流速0.8 mL/min进样量10uL,柱温为室温,检测器波长280nm.流动相A为甲醇,流动相B为1%乙酸水溶液,梯度见表1。
表一
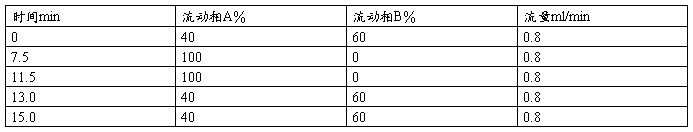
7 分析结果的表述
样品中丁基羟基茴香醚(BHA)、二丁基羟基甲苯(BHT)与特丁基对苯二酚(TBHQ)含量醇测定结果数值以毫克每千克表示(mg/kg).按公式(1)记算:
Xi=Ai×V1×D/V2×1000/M
式中.
Xi——样品中叔丁基羟基茴香醚(BHA)、2,6一二叔丁基对甲酚(BHT)或特丁基对苯二酚(TBHQ)含量的数值.单位为毫克每千克。
Ai——将样品分析所得峰面积代人工作曲线,计算所得进样体积样品中叔丁羟基茴香醚 (BHA)、2.6-二叔丁基对甲酚(BHT)或或特丁基对苯二酚(TBHQ)含量的数值,单位为微克。
V1——加人流动相体积的数位,单位为毫升
V2——进样量的数值,单位为微升。
D一一样液的总稀释倍数。
M一一样品质量的数值,单位为克。
取平行测定结果的算术平均值为测定结果,结果保留一位小数。
(四)花椒里罗丹明的测定
进出口食品中罗丹明B的检测方法( SN/T 2430—2010)
1 范围
本标准规定了进出口食品中罗丹明B的液相色谱检测以及液相色谱—质谱/质谱检测和确证方法。本标准适用于腊鱼、腊肉、香肠、果汁、果酱、辣椒粉、辣椒油、糖果、话梅、葱头及饼干中罗丹明B的测定和确证。
2 方法提要
用乙酸乙酯—环己烷提取试样中的罗丹明B,经凝胶色谱净化系统净化,用液相色谱—荧光检测器或液相色谱—质谱/质谱仪测定和确证,外标峰面积法定量。
3 试剂和材料
除特殊注明外,所有试剂均为分析纯,水为二次蒸馏水。
3.1 甲醇:高效液相色谱级。
3.2 甲酸:高效液相色谱级。
3.3 乙酸乙酯。
3.4 环己烷。
3.5 乙酸乙酯—环己烷(1∶1,体积比)溶液:将乙酸乙酯(3.3)和环己烷(3.4)等体积混合。
3.6 0.2%甲酸:取2.0mL甲酸(3.2),用水定容至1000mL。
3.7 罗丹明B标准品:分子式C28H31CLN203,CAS编号:81-88-9,纯度大于等于99.0%。
3.8 罗丹明B标准储备液:准确称取适量罗丹明B标准品(3.6),用甲醇配制成浓度为100ug/mL的标准储备液。此溶液在0℃~4℃避光保存。
3.9 空白样品提取液:采用空白样品,按照6.1和6.2进行提取和净化后得到的溶液。
3.10 标准工作溶液:根据需要用空白样品提取液将标准储备液稀释成5.0ng/mL、10.0ng/mL、20.0ng/mL、30.0ng/mL、100.0ng/mL的标准工作溶液。置于0℃~4℃避光保存。
3.11 0.22um微孔滤膜,有机系。
4 仪器和设备
4.1 高效液相色谱仪配荧光检测器。
4.2 高效液相色谱—质谱/质谱仪,配电喷雾离子源(ESI)。
4.3 电子天平:感量为0.01g和0.1mg
4.4 凝胶色谱净化系统(GPC)
4.5 组织捣碎机。
4.6 超声波清洗器。
4.7 旋涡混匀器。
4.8 旋转蒸发仪。
4.9 离心机:转速不低于5000r/min。
4.10 具塞塑料离心管:50mL。
5 试样的制备与保存
5.1 试样制备
5.1.1腊鱼、腊肉、香肠、辣椒粉、葱头:取有代表性样约500g,腊鱼、腊肉、香肠需去皮,用组织捣碎机捣碎,装入洁净容器作为试样,密封并做好标识,于-18℃下保存。
5.1.2果酱、辣椒油、饼干:取有代表性样品约500g,搅拌均匀后装入洁净容器内密封并做好标识,于0℃~4℃下保存。
5.1.3话梅:取可食部分约500g,用组织捣碎机捣碎,装入洁净容器作为试样,密封并做好标识,于0℃~4℃下保存。
5.1.4糖果:取约500g糖果,硬糖内层用滤纸,外层用塑料纸包好,然后用锤子捶碎;软糖用剪刀剪碎后,各自混合后装入洁净容器内密封并做好标识,于0℃~4℃下保存。
5.1.5果汁饮料:取有代表性样约500mL,混匀后装入洁净容器内密封并做好标识,于0℃~4℃下保存。
5.2 试样的保存
制样操作过程中应防止样品受到污染或发生残留物含量的变化。
6 测定步骤
6.1 提取
6.1.1腊鱼、腊肉、香肠、果汁、果酱、葱头、糖果、话梅、辣椒粉及饼干
称取2.0g己烷溶液(3.5),于旋涡混匀器上混合提取2min,再超声提取15min后,于4000r/min离心5min,上清液过0.22um微孔滤膜后作待净化液。
6.1.2辣椒油
称取2.0g样品(精确至0.01g)于25mL容量瓶中,加入20mL乙酸乙酯—环己烷溶液(3.5),超声提取15min后,用乙酸乙酯—环己烷溶液(3.5)定容。取15mL溶液过0.22um微孔滤膜后作待净化液。
6.2 净化
取10mL待净化液于GPC样品管中,用于GPC进行净化(参考条件参见附录A中第A.1章),收集洗脱液,于40℃下旋转蒸发至干。残渣用1.0mL甲醇溶解后,过0.22um微孔滤膜,供HPLC—荧光测定,或用1.0uL40%甲醇水定容,过25m微孔滤膜,供HPLC—MS/MS测定或确证。
6.3 测定
6.3.1液相色谱—荧光检测条件
a)色谱柱:C18柱,2.1(内径)×150mm,5um,或相当者;
b)流动相:甲醇—水,梯度洗脱程序见表1;
表1 流动相梯度洗脱程序

c)流速:0.80mL/min;
d)柱温:35℃;
e)进样量:10uL;
f)激发波长(Ex550nm,发射波长(En)580nm。
6.3.2液相色谱—质谱/质谱条件
a)色谱柱:C18柱,2.1mm(内径)×100mm,1.7um或相当者;
b)流动相:甲醇—0.2%甲酸,梯度洗脱程序见表2;
表2流动相梯度洗脱程序
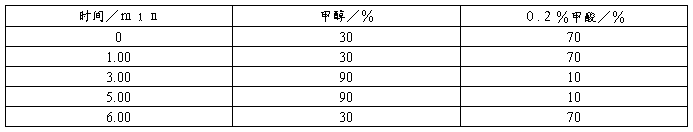
c)流速:0.2mL/min;
d)柱温:35℃;
e)进样量:10μL;
f)离子源:电喷雾离子源;
g)扫描方式:正离子;
h)检测方式:多反应监测(MRM);
6.3.3液相色谱测定
按照6.3.1液相色谱—荧光检测条件对标准工作溶液及样液等体积参插进样测定,样品中待测物含量应在标准曲线范围之内,如果含量超出标准曲线范围,应进行适当稀释后测定。在该条件下,罗丹明B的保留时间约为6.8min。
6.3.4液相色谱—质谱/质谱测定和确证
按照6.3.2液相色谱—质谱/质谱条件测定样液和标准工作溶液,外标标准曲线法测定样液中的罗丹明B含量。样品中待测物含量应在标准曲线范围之内,如果含量超出标准曲线范围,应用空白样品提取液进行适当稀释。在上述色谱条件下,罗丹明B的质量色谱峰保留时间约为4.3min。在相同试验条件下,样品与标准工作液中待测物质的质量色谱峰相对保留时间在2.5%以内,并且在扣除背景后的样品质量色谱图中,所选择的离子对均出现,同时与标准品的相对丰度允许偏差不超过表3规定的范围,则可判断样品中存在对应的被测物。
表3 使用液相色谱—质谱/质谱定性时相对离子丰度最大允许误差

6.3.5空白试验
空白样品按上述测定步骤进行。
7 结果计算和表述
用数据处理软件中的外标法,或绘制标准曲线,按照式(1)计算试样中罗丹明B的含量。
X=(C—Co)*V / *1000…………………………(1)
式中:
X———试样中罗丹明B的含量,单位为毫克每千克(mg/kg);
C———由标准曲线得到的样液中罗丹明B的浓度,单位为纳克每毫升(ng/mL);
Co———由标准曲线得到的空白试验中罗丹明B的浓度,单位为纳克每毫升(ng/mL);
V———样液最终定容体积,单位为毫升(mL);
m———最终样液所代表的试样质量,单位为克(g)。
8 测定低限、回收率
8.1 测定低限
液相色谱—荧光法及液相色谱—质谱/质谱法的测定低限均为0.005mg/kg。
8.2 液相色谱—荧光及液相色谱—质谱/质谱法测定的回收率
(五)测定植物油、酒中的溶剂残留
取5g样品于瓶中,直接上仪器
安捷伦气相色谱仪7890A,后顶空进样器,后FID检测器
(六)测水中的三氯甲烷、四氯化碳
取5g样品于瓶中,直接上仪器
安捷伦气相色谱仪7890A,后顶空进样器,前ECD检测器
(七)测定酒中的塑化剂(领苯二甲酸酯类)
取5g样品于比色管中,再加2毫升正己烷,取上清液1—1.5uL于小瓶中,然后上仪器
气相色谱质谱联用仪。
(八)测定酒中的己酸乙酯
取5g样品于瓶中,直接上仪器
安捷伦气相色谱仪7890A,后顶空进样器,后FID检测器
五、注意事项:
1、ECD检测器不能自行拆解,里面有放射性元素
2、凡事用于测塑化剂的仪器使用过程中不得与塑性材料接触,且清洗后应用丙酮泡,然后烘干,丙酮回收利用。
3、使用色谱柱时,切色谱柱的要求,必须平整,不要接错柱子
4、每次根据进样数量,确保仪器洗液充足。7890A用的是正己烷和蒸馏水作洗液
5、取正己烷用指定的移液管,不得错用,乱用。
六、实习体会
此次实习虽然时间不是很长,但是实习效果很明显,比如说在实习过程中了解了色谱分析的原理和应用、各种色谱仪的结构和操作、内标法和外标法等等。另外,在实习过程中,老师给我们布置任务,实验器材的准备——取样样品处理——实验数据的处理——问题的分析,整个过程全部由我们临时组成的团队独立完成,在这个过程中增强了我们的动手能力、团队协作能力和解决问题的能力,这是我们日后走出社会,走到工作岗位上需要的核心能力,也是我们在课本上永远学不到的能力。
此外,非常感谢姜主任、高老师和连老师的悉心教导以及给予我们生活上的帮助和照顾。