一.药物作用的生物学基础
1.药物在分子水平作用分类:①非特异性结构药物:药理作用与化学结构类型的关系较少,主要受药物理化性(脂水分配系数)质的影响 ②特异性结构药物:发挥药效的本质是药物小分子与受体生物大分子的有效结合,包括立体空间上互补,在电荷分布上相匹配,通过各种键力的作用使二者相互结合,进而引起受体生物大分子构象的改变,出发集体微环境产生与药效有关的一系列生物化学反应。
2.生物靶点:①定义:与药物结合的受体生物大分子 ②种类:受体<例:G-蛋白偶联受体(GPCR)>、酶、离子通道、核酸 ③存在位置:机体靶器官细胞膜上、细胞浆内。
<1>以受体为靶点:药物与受体结合才能产生药效。(治疗高血压的血管紧张素2受体拮抗剂:沙洛坦,依普沙坦;中枢镇痛的阿片受体激动剂:丁丙诺啡,布托啡诺;阿尔法受体激动剂:阿芬他尼。)(受体亚型:肾上腺能受体:α1,α2,α3,β1,β2,β3,多巴胺受体D1,D2,D3,D4,D5,5-羟色胺受体:5-HT1A-1F) 孤儿受体:其编码基因与某一类受体家族成员的编码有同源性,但目前在体内还没有发现其相应的配基
<2>以酶为靶点:由于酶催化生成或灭活一些生理反应的介质和调控剂,因此,酶构成了一类重要的药物作用靶点。(降压药的血管紧张素转化酶抑制剂;肾上腺素抑制剂、调血脂药HMG-CoA还原酶抑制剂;康前列腺增生治疗药物中的5阿尔法还原酶抑制剂;非甾体抗炎药物中的环氧化没(COX—2)抑制剂;抗肿瘤药物中的芳构化酶抑制剂 一氧化氮氧化酶抑制剂)
<3>以离子通道为靶点:(Ⅰ类抗心律失常药为Na+通道阻断剂,主要药物:奎尼丁,利多卡因,美西津,恩卡尼,普罗帕酮;Ca2+拮抗剂:硝苯地平,尼卡地平,尼英地平,帕罗地平,非洛地平;K+通道激活剂:色马凯伦,尼可地尔,吡那地尔)
<4>以核酸为靶点:诺霉素和阿霉素
3.治疗效果:药物在体内发挥作用的关键:①药物到达作用部位的浓度(药物的动力学时相:通常以生物利用度和药代动力学参数来进行描述)②药物与生物的靶点相结合(药效学时相)
4.理化性质对药效的影响:
①溶解度分配系数对药效的影响:脂水分配系数:P=Co/Cw.(正辛醇化学性质稳定,本身无紫外吸收,便于测定药物浓度)
药物化学结构决定其水溶性和脂溶性。水溶性:<1>分子的机型和结合的极性基因<2>形成氢键的能力<3>晶格键
②溶解度对药效的影响:(例:<1>弱酸性药物:巴比妥、水杨酸类,在胃液中几乎不溶解成分子型,易在胃中吸收<2>弱碱型药物如奎宁和麻黄碱在胃液中几乎全为离子型,很难吸收,须在肠中吸收<3>碱性极弱的咖啡因和茶碱,在酸性介质中解离也也很少,在胃内易吸收<4>完全离子化的季铵盐类和磺酸类,脂溶性差,消化道吸收也差,更不容易通过血脑屏障达到脑部)
5.药物立体结构对药效的影响:(几何异构和光学异构对药物活性有较大影响,例如:在雌激素的构效关系研究中,发现两个含氧官能团及氧原子间的距离对生理作用是必须的,而甾体母核对雌激素并非必须结构)
①几何异构是由双键或环等刚性或半刚性系统导致分子内旋转受到限制而产生的,几何异构体的理化性质和生理活动都有较大差异。如顺、反式己烯雌酚例子
②光学异构分子中存在手性中心,两个对映体互为实物和镜像,除了将偏振光向不同的方向旋转外,有着相同的物理性质和化学性质,但其生理活性则有不同的情况
(<1>在有些药物中,光学异构体的药理作用相同,如左旋和右旋氯喹具有相同的抗疟活性,但在很多药物中左旋和右旋的生物活性并不相同<2>光学活性体药物的两个对映体在活性上的表现可有作用完全相同,作用相同但强度不同,作用方式不同等几种类型<3>有时一个对映体能抵消另一对映体的部分药效<4>除了药物受体对药物的光学活性有选择外,由于生物膜,血浆和组织上的受体蛋白和酶,对药物进入机体后的吸收,分布和排泄的过程,均有立体选择性的优先通过与结合的情况导致药效上的的差别)
{D-(-)肾上腺素通过三个集团与受体结合:①氨基②苯环及其两个酚羟基③侧链上的醇羟基}
6.药物——受体相互作用的化学本质:
药物分子和受体的结合,除静电相互作用外,主要是通过各种化学键的连接,形成药物——受体复合物,其中共价键得键很大,结合是不可逆的。
①共价键的结合:这是药物和受体间可以产生的最强的结合键,他难以形成,但一旦形成也不易断裂。共价键是由有关原子间共享电子而形成的,在外部介质中只有当使用加热和活性较大的化学试剂时大部分共价键才能断裂,然而体内生物介质中大多数共价键是在温和的条件下通过酶的催化过程形成和断裂的(Eg:某些有机磷杀虫药,胆碱酯酶抑制剂和烷化剂类抗肿瘤药都是过;具有高张力的四元环内酯或内酰胺类药物如β-内酰胺类抗生素:青霉素的抗菌作用就是由于它能和细菌细胞壁生物合成中的转肽酶生成共价键)
②非共价键的相互作用:<1>他们和相应受体间的结合通常是建立在离子键或更弱的结合力上,这些力于药物受体来说已足够牢固和稳定,使其不太易于从作用部位出去。(在生理ph时,药物分子中的羧基,磺酰胺基和脂肪组氨基等基团,均成电离状态,季铵盐在任何ph下都成电离状态。大多数带电荷的药物为阳离子,少数为阴离子。另一方面,主要由蛋白质构成的受体,其分子表面也有许多可以电离的基团)<例:精氨酸和赖氨酸的碱性基团,在生理ph时全都质子化,生成带正电荷的阳离子;组氨酸的咪唑环,色氨酸的吲哚环也可以质子化,但程度较低;冬氨酸和谷氨酸的酸性基团在生理ph时,通常完全电离,生成阳离子基团>(药物的离子与受体带相反电荷的离子可形成离子键结合。药物——受体间之间形成的这种例子间的结合,是非共价键中最强的一种,是药物受体复合物形成过程中的第一个结合点)<2>非共价键类型:加强的离子键、离子键、离子——偶极、偶极——偶极、氢键、电荷转移、疏水性相互作用、范德华相互作用(当接近到一定程度时,分子的其余部分还能与受体通过分子间普遍存在的范德华引力相互吸引,这样药物与受体就结合形成复合物)
二.新药开发的基本途径与方法
1.新药的药物化学研究通常分为两个阶段:①发现先导化合物②对先导化合物进行优化
2.先导化合物:有独特结构的具有一定活性的化合物
3.先导化合物的发现途径:
①从天然产物中得到<1>植物:(从中药青篙中分离出的抗疟有效成分青篙素为新型结构的倍半萜过氧化物。后采用结构修饰的方法合成了抗虐效果更好的篙甲醚和青篙素琥珀酸脂,治疗效果比青篙素高5倍,毒性低) <2>微生物资源(对β-内酰胺类抗生素特别敏感的菌株,用不同的β-内酰胺酶作区别试验,发现了克拉维酸和沙纳霉素等强力抑制β-内酰胺酶活性的药物) <3>动物内源性物质
②现有的药物作为新药研发的基础:<1>由药物副作用发现先导化合物(毒副作用:药物用于治疗的称治疗作用,其他作用常称为毒副作用)(例:a. 抗组胺药异丙嗪的镇静副作用→吩噻嗪类抗精神失常药;在发现了磺胺利尿的副作用系抑制碳酸酐酶的结果后,先后合成了许多磺胺酰类利尿药,如:呋塞米、吡咯他尼等都有很强的利尿作用)<2>通过药物代谢研究得到先导物(抗抑郁药丙咪嗪和阿米替林的代谢物去甲丙咪嗪和去甲阿米替林,抗抑郁作用比原药强,且副作用小,生效快)<3>以现有突破性药物作先导:例如兰索拉唑及其它的拉唑的研究是以奥美拉唑为先导物的,其活性比奥美拉唑更强(Me-too药物:具有自己知识产权的药物,其药效和同类的突破性的药物相当)
③用活性内源性物质作先导化合物:现代生理学认为,人体被化学信使{生理介质或神经递质所控制。体内存在一个非常复杂的信息交换系统,每一个信使都具各自特殊的功能,并在其作用的特定部位被识别。患病时机体失去了平衡,而药物治疗,就是用外源性的化学物质(信使),来帮助机体恢复平衡}
(合理药物设计:根据对生理病理的了解来研究新药,通常是针对与该生理活动有关的酶或受体来设计新药)
④利用组合化学和高通量筛选得到先导化合物:(组合化学:对含有数十万乃至数十亿个化合物的化学品库进行同步的合成和筛选。也称非合理药物设计)
4.先导化合物优化方法:生物电子等排体替换、前药设计、软药设计、定量构效关系研究
①生物电子等排体:具有相似的物理和化学性质,又能产生相似的生物活性的相同价键的基团
经典电子等排体:<1>一价电子等排体:如卤素和XHn基团,X=C、N、O、S<2>电子等排体:R-O-R’、R-NH-R’、R-CH2-R、R-Si-R’<3>三价电子等排体:-N=、-CH=<4>四价电子等排体:=C=、=N=、=P=
非经典电子等排体:<1>可替代性基团:-CH=CH-、-S-、-O-、-NH-、-CH2-<2>环与非环结构的替代(组胺H2受体拮抗剂中环内等价电子等排体的应用较为成功:呋喃环和噻唑环置换咪唑环得雷尼替丁和法莫替丁)
②前药设计:<1>概念:如果药物经过化学修饰后得到的化合物,在体外没有或很少有活性,在生物体或人体内通过酶的作用又转化为原来的药物而发挥药效时,则称原来的药物为母体药物,修饰后得到的化合物为前体药物,简称前药<2>前药目的a.增加药物的代谢稳定性b.干扰转运特点,使药物定向靶细胞,提高作业选择性c.消除药物的副作用或毒性以及不适气味d.适应剂型的需要(羧苄青霉素口服时对胃酸不稳定,易被胃酸分解失效。将其侧链上的羧基酯化为茚满酯则对酸稳定,可供口服,吸收也得以改善)
③软药设计:<1>硬药:在体内不受任何酶攻击的有效药物,以免有害代谢物产生,实际上“硬药”并未取得应有的效果<2>容易代谢失活的药物,使药物在完成治疗作用后,按预先规定的代谢途径和可以控制的速率分解、失活并迅速排出体外,从而避免药物的蓄积毒性。{阿曲库铵的双季铵结构间由13个原子的链联接。链上有双酯结构,而且在季氮原子的β位含有强吸电子的脂基。阿曲库铵在生理PH和体温下,由于季氮原子的β位上的强吸电子的作用,可进行霍夫曼消除,生成N-甲基四氢罂粟碱和其他代谢物,链上的双酯的也可被血浆中的酯酶水解,这种性质避免了肌肉松弛药蓄积中毒副作用}
④定量构效关系:用数学函数式来表示同类药物结构变化后活性的变化(同类药物:是指具有相同基本结构并有同一药理作用类型的药物)
5.利用定量构效关系方法进行先导物优化的方法
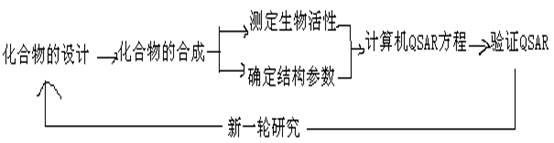
6.常用化学结构参数:电性参数、疏水性参数、立体参数
1.药物化学定义:是一门发现与发明新药、合成化学药物、阐明药物化学性质、研究药物分子与机体细胞(生物大分子)之间互相作用规律的综合性学科,是要学领域中的带头学科
2.药物化学研究内容:既包含着化学科学,又必须涉及到生命科学的内容,既要研究化学药物的化学结构特征、与此相联系的理化性质、稳定性状况,同时又要了解药物进入体内后的生物效应、毒副作用及药物进入体内的生物生物转化等化学——生物内容
3.与药物作用密切相关的因素:①解离常数(Pka)②脂水分配系数③体内代谢过程(①②作用的强弱和快慢③作用的长短)
4.①钠通道阻滞剂:硫酸奎尼丁②钾通道阻滞剂:盐酸胺碘酮③血管紧张素拮抗剂:氯沙坦④强心苷:地高辛
5.缩写:①羟甲戊二酰辅酶A:HMG-CoA②血管紧张素转化酶抑制剂:ACEI③血管紧张素Ⅱ:AngⅡ
6.喹诺酮类抗菌药物的发展概述
①第一代喹诺酮抗菌药物的药效学特征,抗革兰氏阴性菌药物,对革兰氏阳性菌几乎无作用。其活性中等,体内易被代谢,作用时间短,中枢毒性较大,易产生耐药性, 现已少用。(奥索利酸、吡咯米酸 ) ②第二代喹诺酮抗菌药的药效学特征,抗菌活性增强。抗菌谱也从革兰氏阴性菌扩大到阳性菌,并对绿脓杆菌有效,药代动力学性质也得到改善,耐药性低,毒副作用小,临床上用于治疗泌尿道感染和肠道感染及耳鼻喉感染抗革兰氏阴性菌药物,化学结构特征为在分子中的7位引入哌嗪基团(西诺沙星,吡哌酸) ③第三代喹诺酮抗菌药的药效学特征,具有抗革兰氏阳性与阴性菌药物的活性,对支原体、衣原体、军团菌及分枝菌有作用。化学结构特征为在分子中的6位引入氟原子。1位、5位、8位上的取代基改变及7位哌嗪基被其电子等排体替代(环丙沙星)
7.四环素代表药物:金霉素、土霉素、四环素等及半合成衍生物
8.代谢拮抗原理:Wood-Fields学说开辟了从代谢拮抗寻找新药的途径。代谢拮抗是指设计与生物体内基本代谢物的结构具有某种程度相似的化合物,使与基本代谢物竞争性或干扰基本代谢物的利用,或掺入生物大分子的合成之中形成伪生物大分子,导致致死合成,从而影响细胞的生长。抗代谢的设计多采用生物电子等排原理。
9.Vitamin A:抗干眼病、Vitamin A 缺乏症
Vitamin D2、Vitamin D3:预防Vitamin D 缺乏症、佝偻病、骨软化病
Vitamin E:预防Vitamin E 缺乏症、间歇性跛行
Vitamin K1:抗出血维生素,用于新生儿出血症、吸收不良或口服抗凝剂所致的低凝血酶原症,长期应用广谱抗生素所致的 Vitamin K 体内缺乏症
Vitamin B1:Vitamin B 缺乏症、周围神经炎
Vitamin B2:核黄素缺乏症
Vitamin B6:Vitamin B6 依赖综合症、缺乏症、先天性代谢障碍症
Vitamin B12:恶性贫血、巨幼红细胞性贫血、抗叶酸药引起的贫血、神经系统疾病
Vitamin H:用于生物素酶缺乏的儿童
Vitamin C:Vitamin C 缺乏症、酸化尿、特发性高铁血红蛋白症
10.脂溶性维生素:维生素A 醋酸酯、维生素、维生素E 醋酸酯;水溶性维生素:维生素 C、生物素
11. 治疗高血压的血管紧张素2受体拮抗剂:沙洛坦,依普沙坦
中枢镇痛的阿片受体激动剂:丁丙诺啡,布托啡诺
阿尔法受体激动剂:阿芬他尼
Na+通道阻断剂,主要药物:硫酸奎尼丁,利多卡因,美西津,恩卡尼,普罗帕酮
Ca2+拮抗剂:硝苯地平,尼卡地平,尼英地平,帕罗地平,非洛地平
K+通道激活剂:色马凯伦,尼可地尔,吡那地尔
钾通道阻滞剂:盐酸胺碘酮
血管紧张素拮抗剂:氯沙坦
强心苷:地高辛
四环素代表药物:金霉素、土霉素、四环素等及半合成衍生物
第二篇:制药工程(化学制药方向的药物合成实验总结)
一、目的要求
1.熟悉溴化、Delepine反应、乙酰化、羟甲基化、Meerwein-Ponndorf-Verley羰基还原、水解、拆分、二氯乙酰化等反应的原理。
2. 掌握各步反应的基本操作和终点的控制。
3. 熟悉氯霉素及其中间体的立体化学。
4. 了解播种结晶法拆分外消旋体的原理,熟悉操作过程。
5. 掌握利用旋光仪测定光学异构体质量的方法。
二、实验原理
氯霉素的化学名为1R,2-(-)-1-对硝基苯基-2-二氯乙酰胺基-1,3-丙二醇,(1R,2R)-(-)-p-nitropHenyl-2-dichloroacetamido-1,3-propanediol。氯霉素分子中有两个手性碳原子,有四个旋光异构体。化学结构式为:
上面四个异构体中仅1R,2R(-)〔或D(-)苏阿糖型〕有抗菌活性,为临床使用的氯霉素。
氯霉素为白色或微黄色的针状、长片状结晶或结晶性粉末,味苦。mp.149~153℃。易溶于甲醇、乙醇、丙酮或丙二醇中,微溶于水。比旋度〔α〕25-25.5°(乙酸乙酯);〔α〕D25+18.5°~21.5°(无水乙醇)。
合成路线如下:
三、实验方法
(一)对硝基α-溴代苯乙酮的制备
在装有搅拌器、温度计、冷凝管、滴液漏斗的 250 mL四颈瓶中,加入对硝基苯乙酮10 g,氯苯75 mL,于25~28℃搅拌使溶解。从滴液漏斗中滴加溴9.7 g 。首先滴加溴2~3滴,反应液即呈棕红色,10 min内褪成橙色表示反应开始;继续滴加剩余的溴,约1~1.5 h加完,继续搅拌1.5 h,反应温度保持在25~28℃。反应完毕,水泵减压抽去溴化氢约30 min,得对硝基α-溴代苯乙酮氯苯溶液,备用。
注释:
1. 制备氯霉素的二、实验原理除以对硝基苯乙酮为原料的二、实验原理(对酮法)外,还有成肟法、苯乙烯法、肉桂醇法、溴苯乙烯法以及苯丝氨酸法等。
2. 冷凝管口上端装有气体吸收装置,吸收反应中生成的溴化氢。
3. 所用仪器应干燥,试剂均需无水。少量水分将使反应诱导期延长,较多水分甚至导致反应不能进行。
4. 若滴加溴后较长时间不反应,可适当提高温度,但不能超过50℃,当反应开始后要立即降低到规定温度。
5. 滴加溴的速度不宜太快,滴加速度太快及反应温度过高,不仅使溴积聚易逸出,而且还导致二溴化合物的生成。
6. 溴化氢应尽可能除去,以免下步消耗六亚甲基四胺。
思考题:
1. 溴化反应开始时有一段诱导期,使用溴化反应机理说明原因?操作上如何缩短诱导期?
2. 本溴化反应不能遇铁,铁的存在对反应有何影响?
(二)对硝基α-溴化苯乙酮六亚甲基四胺盐的制备
在装有搅拌器、温度计的 250 mL三颈瓶中,依次加入上步制备好的对硝基α-溴代苯乙酮和氯苯20 mL,冷却至15℃以下,在搅拌下加入六亚甲基四胺(乌洛托品)粉末8.5 g,温度控制在28℃以下,加毕,加热至35~36℃,保温反应1 h,测定终点。如反应已到终点,继续在35~36℃反应20 min,即得对硝基α-溴代苯乙酮六亚甲基四胺盐(简称成盐物),然后冷至16~18℃,备用。
注释:
1. 此反应需无水条件,所用仪器及原料需经干燥,若有水分带入,易导致产物分解,生成胶状物。
2. 反应终点测定:取反应液少许,过滤,取滤液1 mL,加入等量4% 六亚甲基四胺氯仿溶液,温热片刻,如不呈混浊,表示反应已经完全。
3. 对硝基α-溴代苯乙酮六亚甲基四胺盐在空气中及干燥时极易分解,因此制成的复盐应立即进行下步反应,不宜超过12 h。
4. 复盐成品:mp.118~120℃(分解)。
思考题:
1. 对硝基-α-溴代苯乙酮与六亚甲基四胺生成的复盐性质如何?
2. 成盐反应终点如何控制?根据是什么?
(三)对硝基-α-氨基苯乙酮盐酸盐的制备
在上步制备的成盐物氯苯溶液中加入精制食盐3 g,浓盐酸17.2 mL,冷至6~12℃,搅拌3~5 min,使成盐物呈颗粒状,待氯苯溶液澄清分层,分出氯苯。立即加入乙醇37.7 mL,搅拌,加热,0.5 h后升温到32~35℃,保温反应5 h。冷至5℃以下,过滤,滤饼转移到烧杯中加水19 mL,在32~36℃搅拌30 min,再冷至-2℃,过滤,用预冷到2~3℃的6 mL乙醇洗涤,抽干,得对硝基-α-氨基苯乙酮盐酸盐(简称水解物),mp.250℃(分解),备用。
注释:
1. 对硝基-α-溴代苯乙酮与六亚甲基四胺(乌洛托品)反应生成季铵盐,然后在酸性条件下水解成对硝基-α-氨基苯乙酮盐酸盐。该反应称Delepine反应。
加入精盐在于减小对硝基-α-氨基苯乙酮盐酸盐的溶解度。
3. 成盐物水解要保持足够的酸度,所以与盐酸的摩尔比应在3以上。用量不仅导致生成醛等副反应(Sommolet反应),而且对硝基-α-氨基苯乙酮游离碱本身亦不稳定,可发生双分子缩合,然后在空气中氧化成紫红色吡嗪化合物。此外,为保持水解液有足够酸度,应先加盐酸后加乙醇,以免生成醛等副反应。
4. 温度过高也易发生副反应,增加醛等副产物的生成。
思考题:
1. 本实验中Delepine反应水解时为什么一定要先加盐酸后加乙醇,如果次序颠倒,结果会怎样?
2. 对硝基-α-氨基苯乙酮盐酸盐是强酸弱碱生成的盐,反应需保持足够的酸度,如果酸度不足对反应有何影响?
(四) 对硝基-α-乙酰胺基苯乙酮的制备
在装有搅拌器、回流冷凝器、温度计和滴液漏斗的 250 mL四颈瓶中,放入上步制得的水解物及水20 mL,搅拌均匀后冷至0~5℃。在搅拌下加入醋酐9 mL。另取40% 的醋酸钠溶液29 mL,用滴液漏斗在
30 min内滴入反应液中,滴加时反应温度不超过15℃。滴毕,升温到14~15℃,搅拌1 h(反应液始终保持在pH 3.5~4.5),再补加醋酐1mL,搅拌10 min,测定终点。如反应已完全,立即过滤,滤饼用冰水搅成糊状,过滤,用饱和碳酸氢钠溶液中和至pH 7.2~7.5,抽滤,再用冰水洗至中性,抽干,得淡黄色结晶(简称乙酰化物),mp.161~163℃。
注释:
1. 该反应需在酸性条件下(pH 3.5~4.5)进行,因此必须先加醋酐,后加醋酸钠溶液,次序不能颠倒。
2. 反应终点测定:取反应液少许,加入NaHCO3中和至碱性,于40~45℃温热30 min,不应呈红色。若反应未达终点,可补加适量的醋酐和醋酸钠继续酰化。
乙酰化物遇光易变红色,应避光保存。
思考题:
1. 乙酰化反应为什么要先加醋酐后加醋酸钠溶液,次序不能颠倒 ?
2. 乙酰化反应终点怎样控制,根据是什么?
(五) 对硝基-α-乙酰胺基-β-羟基苯丙酮的制备
在装有搅拌器、回流冷凝管、温度计的250 mL三颈瓶中,投入乙酰化物及乙醇15 mL,甲醛4.3 mL,搅拌均匀后用少量NaHCO3饱和溶液调pH 7.2~7.5。搅拌下缓慢升温,大约40 min达到32~35℃,再继续升温至36~37℃,直到反应完全。迅速冷却至0℃,过滤,用25 mL冰水分次洗涤,抽滤,干燥得对硝基-α-乙酰胺基-β-羟基苯丙酮(简称缩合物),mp.166~167℃。
注释:
1. 本反应碱性催化的pH值不宜太高,pH 7.2~7.5较适宜。pH过低反应不易进行,pH大于7.8时有可能与两分子甲醛形成双缩合物。
甲醛的用量对反应也有一定影响,如甲醛过量太多,亦有利于双缩合物的形成;用量过少,可导致一分子甲醛与两分子乙酰化物缩合。
为了减少上述副反应,甲醛用量控制在过量40%左右(摩尔比约为1 :1.4)为宜。
2. 反应温度过高也有双缩合物生成,甚至导致产物脱水形成烯烃。
3. 反应终点测定:用玻棒蘸取少许反应液于载玻片上,加水1滴稀释后置显微镜下观察,如仅有羟甲基化合物的方晶而找不到乙酰化物的针晶,即为反应终点(约需3 h)。
思考题:
1. 影响羟甲基化反应的因素有那些?如何控制?
2. 羟甲基化反应为何选用NaHCO3作为碱催化剂?能否用NaOH,为什么?
3. 羟甲基化反应终点如何控制?
(六)异丙醇铝的制备
在装有搅拌器、回流冷凝管、温度计的三颈瓶中依次投入剪碎的铝片2.7 g,无水异丙醇63 mL和无水三氯化铝0.3 g。在油浴上回流加热至铝片全部溶解,冷却到室温,备用。
注释:
1. 所用仪器、试剂均应干燥无水。
2. 回流开始要密切注意反应情况,如反应太剧烈,需撤去油浴,必要时采取适当降温措施。
3. 如果无水异丙醇、无水三氯化铝质量好,铝片剪得较细,反应很快进行,约需1-2 h,即可完成。
(七) DL-苏阿糖型-1-对硝基苯基-2-氨基-1,3-丙二醇的制备
在上步制备异丙醇铝的三颈瓶中加入无水三氯化铝1.35 g,加热到44~46℃,搅拌30 min。降温到30℃,加入缩合物10 g。然后缓慢加热,约30 min内升温到58~60℃,继续反应4 h。冷却到10℃以下,滴加浓盐酸70 mL。滴毕,加热到70~75℃,水解2 h(最后0.5 h加入活性炭脱色),趁热过滤,滤液冷至5℃以下,放置1 h。过滤析出的固体,用少量20% 盐酸(预冷至5℃以下)8 mL洗涤。然后将固体溶于12 mL水中,加热到45℃,滴加15% NaOH溶液到pH 6.5~7.6。过滤,滤液再用15% NaOH调节到pH 8.4~9.3,冷却至5℃以下,放置1 h。抽滤,用少量冰水洗涤,干燥,得DL-苏阿糖型-1-对硝基苯基-2-氨基-1,3-丙二醇(DL-氨基物),mp.143~145℃。
注释:
1. 滴加浓盐酸时温度迅速上升,注意控制温度不超过50℃。滴加浓盐酸促使乙酰化物水解,脱乙酰基,生成DL-氨基物盐酸盐,反应液中盐酸浓度大致在20%以上,此时AL(OH)3形成了可溶性的AlCl3-HCl复合物,而DL-氨基物盐酸盐在50℃以下溶解度小,过滤除去铝盐。
2. 用20% 盐酸洗涤的目的是除去附着在沉淀上的铝盐。
3. 用15% NaOH溶液调节反应液到pH 6.5~7.6,可以使残留的铝盐转变成AL(OH)3絮状沉淀过滤除去。
4. 还原后所得产物除DL-苏阿糖型异构体外,尚有少量DL-赤藓糖型异构体存在。由于后者的碱性较前者强,且含量少,在pH 8.4~9.3时,DL-苏阿糖型异构体游离析出,而DL-赤藓糖型异构体仍留在母液中而分离。
思考题:
1. 制备异丙醇铝的关键有哪些?
2. Meerwein-Ponndorf-Verley还原反应中加入少量AlCl3有何用?
3. 试解释异丙醇铝—异丙醇还原DL-对硝基-α-乙酰胺基-β-羟基苯丙酮主要生成DL-苏阿糖型氨基物的理由。
4. 还原产物1-对硝基苯基-2-乙酰氨基-1,3-丙二醇水解脱乙酰基,为什么用HCl而不用NaOH水解?水解后产物为什么用20 %盐酸洗涤?
5. “氨基醇”盐酸盐碱化时为什么要二次碱化?
(八) D -(-)-1-对硝基苯基-α-氨基-1,3-丙二醇的制备
1. 拆分
在装有搅拌器、温度计的250 mL三颈瓶中投入DL-氨基物5.3 g,L-氨基物2.1 g,DL-氨基物盐酸盐16.5 g和蒸馏水78 mL。搅拌,水浴加热,保持温度在61~63℃反应约20 min,使固体全部溶解。然后缓慢自然冷却至45℃,开始析出结晶。再在70 min内缓慢冷却至29~30℃,迅速抽滤,用热蒸馏水3 mL(70℃)洗涤,抽干,干燥,得微黄色结晶(粗L-氨基物),mp.157~159℃。滤液中再加入DL-氨基物4.2 g,按上法重复操作,得粗D-氨基物。
2. 精制
在100 mL烧杯中加入D-或L-氨基物4.5 g,1 mol / L稀盐酸25 mL。加热到30~35℃使溶解,加活性炭脱色,趁热过滤。滤液用15% NaOH溶液调至pH 9.3,析出结晶。再在30~35℃保温10 min,抽滤,用蒸馏水洗至中性,抽干,干燥,得白色结晶,mp.160~162℃。
3. 旋光测定
取本品2.4 g,精密称定,置100 mL容器中加1 mol / L盐酸(不需标定)至刻度,按照旋光度测定法测定(《中国药典》1995版二部附录38页),应为(+)/(-)1.36º~(+)/(-)1.40º。
根据旋光度计算:含量 % = (100×α) /(2×2.4×29.5)×100%
其中:α= 旋光度
29.5 = 换算系数
2 = 管长为2 dm
2.4 = 样品的百分浓度
注释:
1. DL-氨基物盐酸盐的制备:在250 mL烧杯中放置DL-氨基物30 g,搅拌下加入20% 盐酸39 mL(浓盐酸22 mL,水17 mL)。加毕,置水浴中加热至完全溶解,放置,自然冷却,当有固体析出时不断缓慢搅拌,以免结块。最后冷至5℃,放置1 h,过滤,滤饼用95% 乙醇洗涤,干燥,即得DL-氨基物盐酸盐。
2. 固体必须全溶,否则结晶提前析出。
3. 严格控制降温速度,仔细观察初析点和全析点,正常情况下初析点为45~47℃。
(九)氯霉素的制备
在装有搅拌器、回流冷凝器、温度计的100 mL三颈瓶中,加入D-氨基物4.5 g,甲醇10 mL和二氯乙酸甲酯3 mL。在60~65℃搅拌反应1 h,随后加入活性炭0.2 g,保温脱色3 min,趁热过滤,向滤液中滴加蒸馏水(每分钟约1 mL的速度滴加)至有少量结晶析出时停止加水,稍停片刻,继续加入剩余蒸馏水(共33 mL)。冷至室温,放置30 min,抽滤,滤饼用4 mL蒸馏水洗涤,抽干,105℃干燥,即得氯霉素,mp.149.5~153℃。
注释:
1. 反应必须在无水条件下进行,有水存在时,二氯乙酸甲酯水解成二氯乙酸,与氨基物成盐,影响反应的进行。
2. 二氯乙酰化除用二氯乙酸甲酯作为酰化剂外,二氯乙酸酐、二氯乙酸胺、二氯乙酰氯均可作酰化剂,但用二氯乙酸甲酯成本低,酰化收率高。
3. 二氯乙酸甲酯的质量直接影响产品的质量,如有一氯或三氯乙酸甲酯存在,同样能与氨基物发生酰化反应,形成的副产物带入产品,致使熔点偏低。
4. 二氯乙酸甲酯的用量略多于理论量,以弥补因少量水分水解的损失,保证反应完全。
思考题:
1. 二氯乙酰化反应除用二氯乙酸甲酯外,还可用哪些试剂,生产上为何采用二氯乙酸甲酯?
2. 二氯乙酸甲酯的质量和用量对产物有何影响?
3. 试对我国生产氯霉素的合成路线和其他合成路线作一评价。
(十)结构确证
1. 红外吸收光谱法、标准物TLC对照法。
2. 核磁共振光谱法。
赞同
0
| 评论
