血液病临床分为三大类型:红细胞疾病、白细胞疾病、出血和血栓性疾病。临床上常见的疾病有白血病、再生障碍性贫血、骨髓增生异常综合症、血小板减少症、多发性骨髓瘤、淋巴瘤、骨骼纤维化、血友病、地中海贫血等。
JAK2基因检测
骨髓增殖性疾病(myeloproliferative diseases, MPD)是一组造血干细胞肿瘤增生性疾病。在骨髓细胞普遍增生的基础上有一系或多系细胞尤其突出,呈持续不断的过度增殖和外周血中成熟细胞数量增多为特征。其临床表现具有异质性,但各亚型几乎都伴有白细胞、血小板及巨核细胞增多,后期出现骨髓纤维化和骨髓衰竭,随病程进展部分可转化为其他疾病。其经典的分类主要分为慢性粒细胞白血病(chronic myelogenous leukemia, CML)、真性红细胞增多症(polycythemia vera, PV)、原发性血小板增多症(essential Thrombocythemia, ET)以及原发性骨髓纤维化(primary myelofibrosis, PMF)等。JAK2是酪氨酸蛋白激酶的一种,可以影响基因的转录调节。JAK2基因特定位点的突变将破坏正常JAK2蛋白酪氨酸激酶活性的自我抑制作用,导致造血细胞的异常增生。
JAK2V617F突变发生在65%~97%的真性红细胞增多症(PV)、23%~57%的原发性血小板增多症(ET)及35%~57%的原发性骨髓纤维化(PMF)患者中。根据JAK2突变与骨髓增生性疾病的密切关系,在修订的2008世界卫生组织(WHO)分类系统中,JAK2突变成为慢性骨髓增殖性疾病(MPD)主要的诊断指标。
检测JAK2基因突变,对诊断慢性骨髓增殖性疾病和白血病都有重要意义。华大基因临床检验中心,可以对JAK2基因的多个突变位点进行检测,不仅自动化、精确度高,还可以检测新突变基因。
STR嵌合体检测
造血干细胞移植是目前临床上治疗白血病、骨髓瘤、淋巴瘤、重型再生障碍性贫血和地中海贫血等血液系统疾病的根本手段。根据来源,造血干细胞移植可以分为自体、异体同基因(如双胞胎)、异基因(如父母、同胞或无血缘关系的供者)移植。随着移植技术的进步以及骨髓库的完善,异基因造血干细胞移植在临床上的应用越来越广泛。
异基因干细胞移植的目的是为受者建立供者型的正常造血与免疫功能,以取代原有的异常的造血及免疫系统。为判断移植是否成功并及时地实施免疫抑制治疗和预后,人们需要对移植后受体体内形成的供、受者外周血细胞嵌合体进行检测以观察其变化趋势。
造血细胞嵌合体是异基因造血干细胞移植后供者和受者血细胞共存于受者体内的现象。在造血干细胞移植后,造血嵌合体一般分为3种类型:当供者细胞占受者的骨髓或外周血的比例大于95%时,供者细胞完全植入,称为完全的供者嵌合状态;若移植后,受者细胞仍出现在骨髓或外周血中,供者细胞占25%—95%时,称为混合嵌合状态;若供者细胞占比小于25%,则称为微嵌合状态。供者细胞嵌合率与移植物排斥及疾病的复发呈负相关,因此移植后动态检测嵌合状态对判断移植效果、实施临床干预尤为重要。
如今,嵌合体的监测已成为同种异基因造血干细胞移植术后患者的常规检测项目,检测方法已由传统的细胞学和遗传学方法发展到分子生物学方法。STR基因检测法敏感性高,可靠性好,定量精确,已成为目前国际骨髓移植登记处(IBMTR)推荐的检测供受嵌合状态的“金标准”。
STR,即短串联重复序列(short tandem repeat,STR),也称做微卫星DNA,是由几个碱基对作为核心单位串联重复形成的DNA 序列。STR广泛存在于真核生物基因组中,具有高度的多态性,按孟德尔规律呈共显性遗传,是目前最理想的DNA遗传标记。在医学亲子鉴定中已经得到广泛应用。
华大基因临床检验中心是国内少数几家掌握STR检测技术的临床检验中心。采用该技术,可以及时准确地动态观察嵌合体的变化,为早期预测移植排斥和白血病复发提供依据;还可以准确判断出复发是供者型还是受者型,为临床治疗提供参考。
白血病相关融合基因定量检测
白血病(leukemia)属于造血系统的恶性肿瘤,是一组高度异质性的恶性血液病,其特点为白血病细胞呈现异常增生伴分化成熟障碍。临床出现不同程度的贫血、出血、发热及肝脾、淋巴结肿大,可危及生命。
白血病融合基因(fusion gene),是白血病的分子生物学特异性标志。近年来,由于分子生物学技术的发展,对白血病细胞分子遗传学改变的了解也不断深入。迄今报道白血病涉及至少数十种融合基因。已经认识到大部分的白血病中存在着染色体结构畸变,包括缺失、重复、倒位、易位等,导致原癌基因及抑癌基因结构变异,原癌基因激活或抑癌基因失活,产生新的融合基因,编码融合蛋白。有些基因是调控细胞增殖、分化和凋亡的转录因子,当基因发生变异,直接影响了下游信号传递途径,导致细胞增殖能力增强、凋亡障碍,分化障碍等,产生白血病表型。
一些典型的白血病融合基因是某种白血病的特异性分子诊断标志,如BCR-ABL融合基因,可出现在95%以上的慢性粒细胞白血病(CML)。患者预后效果的好坏,与融合基因的类型有一定关系,如急性早幼粒细胞白血病(APL)特有的PML-RARa融合基因,对APL患者用全反式维甲酸(ATRA)诱导缓解治疗,其预后非常好,复发率低。而有些基因,如MLL相关融合基因,预后差,死亡率高。
白血病微小残留病,是指在白血病经诱导化疗获完全缓解后或是骨髓移植治疗后,体内仍残留有少量白血病细胞的状态。这些残存的白血病细胞将可能成为白血病复发的根源。白血病一旦复发,病情将更加严重。所以,定期检测白血病微小残留病很有必要。通过检测微小残留病融合基因表达水平,可更早预测白血病的复发;指导白血病的临床治疗,根据融合基因表达水平的多少,决定是否继续化疗;有利于评价药物治疗效果,是否耐药,并依此指导临床更换治疗方案;评价造血干细胞移植的净化效果。
华大基因临床检验中心采用实时荧光定量PCR基因检测法检测白血病的相关融合基因,灵敏度高、特异性强,可以为白血病的诊断、分型、临床治疗和预后判断提供可靠的依据。
人类白细胞抗原(HLA)高分辨基因分型检测
目前我国约有400万名患者等待造血干细胞移植,同时每年新发病例4万人左右。造血干细胞移植是白血病患者、再生障碍性贫血、地中海贫血、重症放射病等人体造血系统及免疫系统严重疾病患者彻底治愈的唯一希望。
而在移植过程中,人类白细胞抗原(Human Leukocyte Antigen,HLA)是决定移植排斥反应高低的重要因素。在进行骨髓和其它器官移植时,供者和受者之间HLA越接近,排斥反应的发生率就越低,移植成功率和移植器官长期存活率就越高;反之,就越容易发生排斥反应。病人同胞间HLA相合的可能性为四分之一,而非血缘关系为1/5000-1/10000,对于一些罕见的HLA类型的患者,配型成功的几率为几万甚至几十万分之一。而我国大部分是独生子女家庭,在骨髓库中寻找与患者HLA匹配的志愿者,成为发现供者的主要途径。
HLA的命名含有四组数字,代表HLA基因的不同部分,如HLA-A*01:01:01:02。第一组数字表示HLA低分辨基因分型,第二组及以上的数字表示高分辨分型。正如下图,HLA的低分辨只能识别HLA的抗原特异性,就像在地图上看天安门,只识其大概;而高分辨则可以在基因水平上准确识别HLA,如同看天安门的高清图,可以确切地看到细节。

目前我国骨髓库(即中华骨髓库,全称中国造血干细胞捐献者资料库)中的HLA分型数据多数是低分辨的,并不能确保供者和患者的HLA真正匹配,患者首先需要在骨髓库中筛选出低分辨配型匹配的志愿者,再和这多个低分辨匹配的志愿者逐一进行高分辨复核,才有可能找到真正合适的供者。有的患者与数十个低分辨匹配的志愿者进行复核后,发现他们均不是合适的供者,甚至有的患者始终找不到匹配的供者,只能在HLA部分匹配的情况下就进行造血干细胞移植,导致术后出现严重的排斥反应,需要服用大量药物来维持生命。
即使最后能够找到较为适合的供者,在低配筛选和多次高配复核的过程中,患者的维持费用和多次低配和高配的配型费用就是一个庞大的数目。更为重要的是,先低配筛选再逐一
高配复核的方法,消耗了大量的时间,很多患者等不到配型成功就已经因并发症而离开人世。因此,推广HLA高分辨配型可以从根本上提高骨髓配型的效率和准确率。如果骨髓库全部“高分入库”,那么查找匹配骨髓的时间将有可能从几个月缩短为几分钟——患者的HLA高分数据数据直接与骨髓库中的高分数据进行比对,一步到位找出供者。HLA高分辨分型技术的推广不仅将为患者节约多次低配费用的额外负担,更重要的是,为患者争取到了大量“救命”的时间。
华大基因是中华骨髓库的合作伙伴,承担了国内和国际上大量的HLA配型检测任务。目前,华大基因应用新一代的测序技术,只需通过一次实验就能够读取数千份样本的HLA序列数据,并一次性达到HLA分型的高分辨率,同时还可发现新的等位基因。在检测通量、数据质量和成本控制等方面都有质的飞跃。20xx年,华大基因保质保量地完成了中华骨髓库高分辨入库项目,样本结果质评准确率达到99.75%(骨髓库准确率标准为97%),真正实现效率和准确率的双赢。
华大基因的HLA高分辨分型技术,使建立高分辨HLA数据库成为现实。这将帮助广大患者快速准确地找到合适的供者,也将极大地提高骨髓库的使用率,还可以为HLA的科学研究与技术创新提供基础性的数据支持。
人类血小板抗原(HPA)分型
血小板是血液中的有形成分之一,在止血及维持血管内皮细胞的完整性方面具有重要作用。目前,血小板输注已成为肿瘤、恶性血液病、骨髓衰竭等疾病及外周造血干细胞移植患者治疗的一个重要治疗手段。
HPA即人类血小板抗原,血液供者和受者的HPA不相合可导致血小板输注无效(PTR)、输血后紫癜(PTP)以及新生儿同种免疫性血小板减少性紫癜(NAITP)等。因此,建立准确的HPA分型技术对于血小板减少症的诊断和血小板输注等临床医疗活动具有非常重要的意义。
一直以来,医学上一直用传统的血清学方法进行HPA分型,由于缺乏高质量、高效价抗血清等多种因素的制约,始终无法对HPA进行全面准确的分型。随着对HPA基因的深入研究,将基因检测技术应用于血小板分型中,极大地提高了HPA分型的水平。
华大基因临床检验中心(简称华大临检中心)掌握了多种HPA基因分型方法,可以满足不同的分型需求。华大临检中心提供的HPA基因检测服务不仅通量高,适合大量样本的规模检测;而且结果稳定;并能够检测出18个血小板抗原系统。华大临检中心的HLA分型技术
已经非常成熟,患者在进行血小板输注前,可以进行HPA联合HLA分型检测,更具可靠性和和安全性;而且中心对HPA进行高通量检测,在成本、准确性上比起其他机构更具优势。 华大临检中心提供的HPA分型检测结果,不仅可以作为血小板减少症的诊断和血小板输注的依据;还可以在更大范围里建立血小板数据库,一方面可以让受者找到与之匹配的供者,及时进行疾病治疗;另一方面可以更好的研究HPA在中国人群中分布的基因频率,给HPA相关疾病研究提供依据。
Kir PCR-SSP分型检测
Kir (The killer cell immunoglobulin-like receptor),即杀伤细胞免疫球蛋白样受体,在病毒感染、肿瘤形成、移植受者免疫状态和自身免疫性疾病中发挥着重要作用。
在造血干细胞移植中,供者和受者的HLA(人类白细胞抗原)相合程度越高,移植就越容易成功;而Kir则相反,研究表明,供者和受者的Kir不相合,会降低排斥的发生风险。因此Kir分型可以为骨髓移植供受者的选择提供参考,有效的提高骨髓移植成功率。
华大基因临床检验中心采用PCR-SSP技术对kir进行分型检测,不仅简单、快速、低成本,而且准确性高,可以成为供临床治疗参考的可靠依据。
地中海贫血基因检测 地中海贫血(俗称“软癌”)是一种遗传性溶血性贫血,是由珠蛋白基因的缺陷导致血红蛋白组成成分发生改变。地中海贫血,按照血红蛋白受损伤的位置,主要有α地中海贫血(构成血红蛋白的氨基酸的α链受到损伤)和β地中海贫血(构成血红蛋白的氨基酸的β链受到损伤)。也可以按照基因缺损的数目来分为轻型,中间型和重型地中海贫血。
据世界卫生组织和国际地中海贫血协会资料,全世界大约7%的人群携带地中海贫血基因,每年有50万重型地中海贫血新生儿出生。在我国,地中海贫血主要发生在长江流域以南的广东、广西、四川、云南、贵州、湖南、江西、浙江、福建、海南等地,其中两广、云贵以及海南是高发区。我国人群的地贫基因携带率为1.92%~14.95%,其中广东、广西、海南省人群中“地贫”基因携带者占本省总人口的11%至25%,每年有数千重型地贫患儿出生。 若夫妻为同型地中海贫血的携带者,每次怀孕,其子女有1/4的机会为正常,1/2的机会为带因者,另1/4的机会为重型地中海型贫血患者,因此,在遗传咨询及产前诊断方面,这是非常重要的疾病。
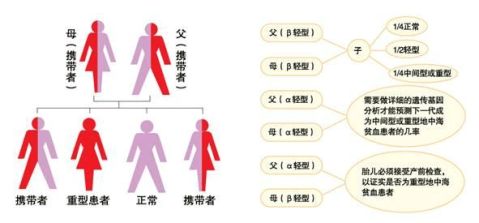
地中海贫血的症状有
1.重型:
出生数日即出现贫血,肝脾肿大进行性加重,黄疸,发育不良,头大,眼距宽,马鞍鼻,前额两颊突出,少数患者在肋骨和脊椎之间发生胸腔肿块,亦可见胆石症,下肢溃疡,常见并发症有急性心包炎,继发性脾功能亢进,血色病。
2.中间型:轻度至中度贫血,患者大多可存活至成年。
3.轻型:轻度贫血或无症状,一般在调查家族史时被发现。
对于重型地贫,目前除造血干细胞移植外无有效根治手段。重型地贫患儿需终身接受定期输血和去铁治疗,每年输血及治疗费用约5万元/人,并随着年龄增长费用逐渐增加。目前造血干细胞移植是根治地贫的唯一手段,移植费用约30万元。
因此,在产前对地中海贫血进行筛查显得十分有必要。控制地中海贫血,预防是关键,婚检、孕检和产检是降低地贫儿出生率的三道防线。
目前地贫有多种筛查方法,但基因检测是诊断地贫的金标准,基因检测能够大大提高地贫检测的准确性,减少地贫的漏检率,从而降低重型地贫的发病率。
第二篇:华大基因广告

动植物重测序五大研究方案
2013/08/01
随着越来越多的物种基因组被破译,重测序的应用也日趋广泛。历时半年,华大科技科研团队根据动植物群体样本类型以及研究领域特点,并融合多年的项目经验,隆重推出重测序研究五大方案,为您提供系统全面的一站式服务,助力项目申请和高水平文章的发表!
方案一:育成动植物驯化基因挖掘方案
研究目的:
针对不同的品种/品系,通过群体内pooling建库的方法,进行全基因组重测序(5X/群体),采用生物信息学方法全基因组范围内扫描变异位点,并进行选择性清除分析(Selective Sweeps),结合相关区域的基因功能注释信息,挖掘驯化相关的基因,剖析家养动植物的驯化条件、驯化过程及其进化动力。
技术路线:
研究方案:
样本量选择: 家养动植物各品系,及其现存祖先种(每个品系可以选择>10个个体等量DNA混合pooling); 测序策略选择: 各品系多个个体pooling DNA 样品进行PE101测序;
测序深度选择: 全基因组重测序>5X coverage /品系。
适用范围:
驯化动物:如家鹅、家鹅、家鸭、家猫、家牛等;
栽培植物:如水稻、玉米、小麦、大豆等粮食作物;苹果、梨、苜蓿、葡萄等经济作物。
方案特色:
1、 高效快速:不需要作图群体,只需要驯化动植物的现有各品种的DNA样本;
2、 成本降低:通过将同一品系中的多个个体(10个左右)进行pooling测序,在兼顾品系代表性的同时最大限度地降低了测序量;
3、 可行性高:目前,运用该思路成功完成驯化研究的动物包括家蚕、家鸡和家犬。
经典案例:
? 样品选取:
– 分布于世界各地的不同品系的狼和犬,其中狼7个品系,犬14个品系
? 测序策略 – 对12只狼的DNA样本进行pooling测序,深度为6.2X
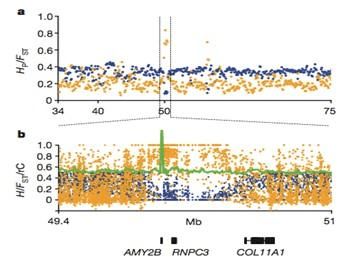
;对60只犬的DNA样本进行pooling测序,测序深度为29.8X
? 分析结果
– 识别了380万个遗传变异位点;
– 在犬的基因组中共发现36个明显受驯化选择的区域,包含122个基因;
– 发现19个与脑发育相关的基因、3个与精子受精过程竞争相关的基因、10个与淀粉和脂肪代谢相关的基因。 参考文献:
[1]Rubin, C. J. et al. Whole-genome resequencing reveals loci under selection during chicken domestication. Nature 464,587-591;
[2]Leonard, J. A. et al. Ancient DNA evidence for Old World origin of New World dogs. Science 298, 1613-1616.
方案二:eQTL方案
研究目的:
通过测序获得各基因型的表达数据,并作为一个数量性状,进行基因表达的数量定位分析(eQTL),进而寻找控制基因表达的上游调控位点,挖掘受该基因调节的下游基因及与该基因协同作用的基因,并建立基因调控网络,从而在表达及调控两个水平上研究控制复杂性状的遗传基础。

技术路线:
研究方案:
样本量选择: 分离群体(DH, RIL, F2,F3 ,BC 等),建议样本数100 个以上。 测序策略选择:
有参考基因组:芯片测序/重测序(3-5X/样品)+RNA-Seq(5M clean reads/样品);
无参考基因组:转录组测序(2-4G clean data /样品);
项目周期: 样本个数≤100,需90 个工作日;样本个数>100,需根据实际项目情况而定。 适用范围:
拥有作图群体的动植物。
方案特色:
1、 更精细的遗传连锁图谱(若分子标记连锁图谱未知);
2、 更精确的作物农艺、经济性状(或目标性状)相关候选基因定位;
3、 更快捷的基因表达调控网络构建;
4、 可与其他结果或方案(如QTL结果,GWAS方案等)相互衔接。
经典案例:
? 样品选取
– 368份玉米自交系(3个作图群体:Illinois high-oil 群体,Alexho 单籽粒合成群体,北京高油群体)
? 测序策略 – 对样本分别进行转录组测序,平均每个样品6.6 Gb raw data,获得103万个SNP;使用Illumina MaizeSNP50 BeadChip 获得56110个SNP,再将两部分SNP数据整合用于后续分析。
? 分析结果 – 利用RNA-seq 测序以及Illumina MaizeSNP50 BeadChip 的办法获得了一百多万个单核苷酸多态性(SNP)位点;
– 利用全基因组关联分析的方法,对籽粒油份相关性状进行了分析,共发现74个基因(loci)与籽粒油份显著关联,其中三分之一是编码油脂代谢的关键酶基因;
– 发现26个与玉米籽粒总含油量显著相关的基因,可以解释总油份83%的表型变异,为玉米油合成的遗传机制提供了视角,并有助于高油玉米的分子育种。
参考文献:
[1] Li H, Peng Z, Yang X, Wang W, et al. Genome-wide association study dissects the genetic architecture of oil biosynthesis in maize kernels.Nat Genet. 2013 Jan;45(1):43-50
[2] West MAL, Kim K, Kliebenstein DJ, et al. Global eQTL mapping reveals the complex genetic architecture of transcript-level variation in Arabidopsis. Genetics, 2007, 175(3):1441?1450.
方案三:野生动物种群历史研究方案
研究目的:
基于第二代高通量测序技术,对于有参考序列的物种,可通过全基因组重测序的方法,获得大量变异信息,讨论群体的遗传结构、影响群体遗传平衡的因素以及物种形成的机制,从而探讨野生动物种群演化机制及野生动物濒危的可能原因,为野生动物资源的保护提供重要的理论依据。

技术路线:
研究方案:
样本选择:群体个数建议大于30个。物种内亚群的划分要比较明确,相同亚群内的个体要有一定的代表性。 测序策略选择:群体测序深度建议5-10X;较高的测序深度,可以保证后续挖掘信息的准确性。 项目周期:项目的周期以项目规模大小而定。样本量小于50个,每个样本5X 基因组覆盖度的数据,项目的运转周期为60个工作日;样本数量更多或单个样本覆盖度更高时,项目的运转周期需根据实际项目情况而定。 适用范围:
1、 珍稀的野生动物,尤其是濒危动物个体或群体;
2、 研究与人类生活紧密关联的动物在野生环境下的生活状况和种群变化;
3、 研究昆虫或鸟类在迁徙过程中产生的种群历史演化。
方案特色:
1、 高效性:执行周期短,高通量测序能在短时间内完成多样本测序;
2、 检测范围广:能够检测到全基因组范围的变异信息,即使是低频信息也能被找到;
3、 信息挖掘全面性:信息分析内容丰富,包含群体结构分析、进化树分析、连锁不平衡分析、主成分分析等群体相关信息;
4、 针对性强:针对野生动物、迁徙类动物群体演化关系,设计一系列研究方法,解决种群历史变化的问题。 经典案例:
? 样品选取:
– 来自六大山系的34只野生大熊猫;其中,秦岭山系8只、岷山山系7只、邛崃山山系15只、大相岭山系1只、小相岭山系1只、凉山山系2只。
? 测序策略选择:
– 进行全基因组重测序,每个个体测序深度为4.7X,个体数据量为10.5Gb。
? 分析结果: – 在这项研究中,研究人员对34个野生大熊猫进行了全基因组重测序,发现当前的6个熊猫地理种群可以分为三个遗传系,包括秦岭(QIN)、岷山(MIN)和邛崃山-大小相岭-凉山(QXL);
– 通过重建熊猫的种群史,研究人员发现了几个重要的进化事件,例如两次种群扩张、两次瓶颈和两次种群分化;
– 研究结果表明,全球气候变化是上百万年来大熊猫种群波动的主要因素,人类活动有可能是近期熊猫种群分化和数量严重下降的重要原因。
参考文献:
[1] Zhao S, Zheng P, Dong S, et al. Whole-genome sequencing of giant pandas provides insights into demographic history and local adaptation. Nat Genet. 2013 Jan;45(1):67-71.
[2] Zhang B W, et al. Genetic viability and populationhistory of the giant panda, putting an end to theEvolutionary Dead End.Mol. Biol. Evol.,2007, 24:1 801-1810.
方案四:动植物目标性状GWAS研究方案
研究目的:
通过全基因组大样本低深度重测序、全基因组芯片分型及大样本简化基因组测序三种策略对动植物重要种质资源进行全基因组的基因型鉴定,并与关注的表型数据进行全基因组关联分析(GWAS),找出与关注表型相关的SNP位点,定位数量性状基因。与数量性状相关基因紧密连锁的SNP标记,后续可用于分子标记辅助育种,增快育种进程。
技术路线:

研究方案:
样本量选择:自然群体大小至少300个样品,选取的个体有代表性。样本间不能有明显的亚群分化(例如生殖隔离等);样本的多态性广;研究的表型性状建议选择几个比较重要的表型性状作为研究的重点,不宜过多。 测序策略选择:
1) 基于全基因组低深度重测序的GWAS研究:全基因组重测序,每个样本>5X测序深度;
2) 基于大样本量的简化基因组测序的GWAS研究:简化基因组(RAD/GBS)测序;
3) 基于全基因组芯片分型的GWAS研究:推荐选择>10k SNP位点数的芯片。
适用范围:
动植物自然群体如粮食作物、家禽家畜种质资源。
方案特色:
1、 标记密度高;
2、 无需构建作图群体,自然群体/种质资源都可作为研究材料;
3、 可以一次性同时考察多个性状;
4、 定位更精确;
5、 性状关联位点的贡献率高,应用前景好。
经典案例
:
? 样品选取:
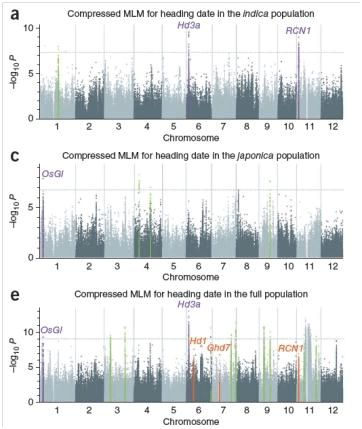
– 950份代表性中国水稻地方品种和国际水稻品种
? 测序策略
– 每个样品平均约1X低深度全基因组重测序
? 分析结果 – 通过GWAS鉴定出32个新的与抽穗期和产量性状相关的变异位点,鉴定出18个候选基因。
参考文献:
[1] Huang X, Zhao Y, Wei X, Li C. Genome wide association study of flowering time and grain yield traits in a world-wide collection of rice germplasm. Nat. Genet, 2011.
[2] Morris GP, et al. Population genomic and genome-wide association studies of agroclimatic traits in sorghum. PNAS, 2013, 110(2):453–458.
方案五:动植物单倍型研究方案
研究目的:
单体型图即HapMap(Haploid Map)是建立存储某一物种常见SNP变异以及LD值等相关信息的数据库。从常见SNP中挑选出更具代表性的标签SNP(Tag SNP),利用这些相对数据量较少的标签SNP集合所包含的基因型信息,就可以代表整个基因组的大部分遗传信息。因此,HapMap的建立,可获得用于设计高密度SNP基因分型芯片的数据库,将大大地简化该物种后续遗传学研究的数据量,从而提高后续相关研究的速度与效率。 技术路线:

研究方案: 样本量选择:已有参考基因组的物种,选择不同地域、不同品种、具有代表性的个体,样本量100个左右。 测序策略选择:PE91测序
测序深度选择:全基因组重测序5-20X /样本
适用范围:
已有参考基因组重要作物与家禽家畜等:如青稞、芝麻、梨、梅花、大豆、谷子、猪、马、牛、羊、鸡、鸭、鹅等。 方案特色:
通过全基因组测序的方法获得物种的多态性SNP以及该物种LD情况,构建单倍型图谱。利用检测到的tag SNP与表型关联进行全基因组关联分析(GWAS)定位QTL;或是设计全基因组分型芯片,用于大群体的基因分型。 经典案例:
? 样品选取:
– 103株玉米(包括驯化前与驯化品系和一个代表性的近缘属Tripsacum)
? 测序策略
– 每个样品约4.2X的全基因组测序数据
? 分析结果
– 构建了一张含5500万个SNP位点的第二代玉米HapMap;
– 发现染色体结的存在或缺失造成了“种内”玉米基因组大小出现很大的差异;种间玉米基因组大小的变化主要与大量的转座子有关;
– 综合利用玉米两代HapMap的标记数据对5个重要的农艺性状进行GWAS分析,与HapMap1结果保持一致,且关联更紧密。
参考文献:
[1]The International HapMap Consortium. A Haplotype Map of the Human Genome. Nature 437, 1299-1320. 2005.
[2]The International HapMap Consortium. Integrating common and rare genetic variation in diverse human populations. Nature 467, 52-58. 2010.
[3]Gore, M.A. et al. A first-generation haplotype map of maize. Science 326, 1115–1117 (2009).