一、混合基组
#p b3lyp/GenECP opt Gen 自己在输入文件末尾定义基组,ECP 使用赝势
空行
g03 opt
空行
0 1
... ...
... ...
空行
C H O 0 碳 氢 氧 数字0 GEN
6-31G** 基组名
**** 结束符号
Ru P Cl 0 Ru P Cl 数字0 GEN
Lanl2dz 基组名
**** 结束符号
空行
Ru P Cl 0 Ru P Cl 数字0 ECP
Lanl2dz 基组
=====================
建议你查看 GEN 关键字。上述,如不加ECP亦可。
二、出现错误提醒:
(一)出现名言提示,表明是正常结束的;l9999终止,是因为循环次数不够。建议用optcyc=100-999增加循环;或者把输出结果做为输入结构再次优化!
1、是我感觉你要是maxcycle=200还是999死的话,就减小一下步长试试,可能会有用的!maxstep=1
2、该先看下收敛指标是不是很接近了,如果很接近就继续做(读chk)或取出最后一个坐标继续算,如果相差很远,可以加非线形实验下,再一个就是加大循环,缩短步长。
(二)L202是重新定位坐标,计算对称性,检查变量的,它出错应该就是输入结构有问题。
三、过渡态
寻找过渡态的时候,可以用TS,QST2 和QST3 方法,那一种方法的效果更好一些?” 取决于计算前所拥有的信息量。当然,信息越多,结果越有把握。所以,QST3 最有把握。如果没有任何初始信息,那就没有什么选择了。“我想不同的输入方法肯定不会产生一样的结果,那输入过程中限制它们间相对位置的条件是什么呢?” 如果输入不同会导致结果不同,那么应当全面考虑。首先可以用化学知识筛选。如果此后仍有多种可能性,就应当通过计算比较。
我算过的用HF方法虚频超过1000,而用DFT只要100-300.所以说方法很重要。但是,只要你找到的TS虚频与反应的方向一致并且IRC已经验证,这个TS就是可信的。虚频也不能太小,比如小于100.
过渡态与你的产物相差甚远 说明在这条反应路径上还有过渡态你没有找到
在优化过渡态的时候圈数设为100,步长5,怎么输入
opt(TS,calcFC,maxcyc=100,maxstep=5)是这样
寻找过渡态不是一件容易的事(对于我和大多数刚涉及量化的人来说),因此我希望通过写这个经验小结能对大家有些帮助。
1.首先遇到的问题是,用哪种方法来寻找过渡态?
GAUSSIAN提供的方法是QSTN和TSN方法。两种方法各有优点和缺点。QSTN方法特别QST3方法要求输入反应物,过渡态的猜测结构,产物这三者的结构。特别麻烦。但很管用,一般不会出现不收敛的情况。对于TSN(对应关键词为OPT=TS)方法,只要求输入过渡态的初始结构,但这个初始结构非常的关键,如果结构不好,则很容易出现不收敛的情况。所以我建议,如果是刚开始做过度态的话,用QSTN方法是好的选择,等有了“感觉”之后,再用TSN方法。
2. 怎么解决经常出现的错误?
在找过度态的时候,经常碰到的一些问题就是(1)不收敛,(2)有一个错误的本征值(错误信息为:there is a wrong sign eigenvalue in hessian matrix.....),(3)和LINK9999错误导致退出。对于不收敛的情况,可以分为两类,比如提示信息里的CONVERGENCE FAILER 提醒收敛到了10(-5),而此时你设定的SCF循环次数也仅仅是64步,那么完全有希望通过加大SCF循环次数来达到收敛的目的。倘若只收敛到10(-3)或10(-2),此时加大循环次数可能就没用了。结果还是CONVERGE FAILER。此时可采用SCF=QC,来达到强制收敛的目的 。因为SCF=QC(LINK508)的计算量比默认的L502要大,所以不到万不得以就不用它了。出现第二个错误可以直接用关键词OPT=NOEIGEN 来实现。LINK9999出错是因为已经走完了默认的步数,但还未完成。系统会自动跳出。出现这种情况大多数就是因为优化步数和SCF步数超过了默认值。可用OPT(MAXCYCLE=100)和SCF(MAXCYCLE=300)来改错。
3.怎么样控制过渡态的优化,使得过渡态不至于收敛到其他的分子结构中去?
我用GAUSS VIEW 可以解决这个问题 ,当刚开始运行GAUSSIAN时,你用GVIEW去打开输出文件时,你可以看到你的过渡态的初始输入结构,当一个循环过后(从上一个LINK502到下一个LINK502),你再打开输出文件,你就可以清晰地看到优化一步后分子的构型,这样就可以随时监控过度态分子的结构,倘若已经有收敛到其他分子构型的趋势时,你就可以把它给KILL了,而不至于需要等全部工作结束后,打开输出文件才知道已经不是想要的过渡态了。 如果收敛到其它的构型上去,可以考虑缩小OPT的步长.iop(1/8=2或3)即可。
4. 还需要加其它的关键词吗?
建议在OPT中加入CALCFC。这样可以加大找到过渡态的几率。本人深有体会!
5. 如何控制寻找过渡态的步长?
这个可以用IOP来实现。具体相应的IOP是iop(1/8)=2,此语句说明在找过度态的时候,以2A(A为基本单位长度)为单位来寻找过渡态。
6.我用高斯03做了两个稳定点A和B的过渡态TS,发现频率文件的振动方向还不足以证明TS就是A和B的过渡态,所以,我又接着做了IRC,结果发现IRC正方向走出的结构与A结构差别很小,而反方向走出的结构与B相差太大,是不是说明这个求出的TS不是AB的过渡态?
那就应该不是,可是你的这个ts也是反映路径当中的一个ts,他前后走出的东西也是im,你如果想要得出AB之间的过渡态的话,最好就用qst2和qst3方法找.
四、奥奥的输入文件
%mem=8mw
%rwf=1,2000mb,2,2000mb,3,2000mb
%chk=ts-1+
#P B3LYP/6-311G(d) fopt=(ts,Z-matrix,calcfc) optcyc=90
iop(5/13=1) iop(9/6=150) iop(1/11=1) freq
**********
1 2
C
S,1,r1
X,1,1.,2,a1
Si,1,r2,3,a1,2,180.,0
Variables:
r1=2.85080391
r2=1.88789137
a1=21.74742362
iop(5/13=1) iop(9/6=150) iop(1/11=1)
吉大ADF 20:54:02
第一个是优化用的 第三个是找过渡态用的
吉大ADF 20:54:15
第二也可以不加
吉大ADF 20:54:44
比较没有第一个 你就是基本的优化都会出问题
ComputerGirl 20:54:55
这几个字段是连在一起用的吗?还是做优化用一个,做过渡态用另一个。
吉大ADF 20:55:18
优化稳定体时 第一个就行
吉大ADF 20:55:29
过渡态时 第一个和第三个
吉大ADF 20:55:33
第二个 不用加 第二个 算能量时会用到
Image point= NEB
五、IRC计算
程老师:
IRC=(forward 或backward stepsize=n) n是个自然数,一般可以为6.
一定要有你算过渡态的CHK文件啊!是从CHK文件读数据的。不是有混合基组的话,如果有多个虚频,说明未找到能力最高点。有机金属配合物很复杂,又使用混合基组,所以情况很复杂,要靠IRC内禀反应坐标扫描了。在我看来,这样的计算就未必准。结果非常依赖基组的大小。DFT那是半经验方法哦~~ 在断裂键和生成键上家极化函数好一些,也好不到哪去。不必了。这种方法用频率验证意义不大
程老师:
可以进行频率验证的,过渡态一般有且只有一个虚频(负频),大小不一定,与要断裂键或新生成键的键长有关系。
程老师 14:14:01
一般要IRC扫描确定过渡态。如果熟练的话,可以不用IRC扫描即可确定过渡态\
宏师姐 20:27:35
算完后要导出坐标,在过渡态的基础上算频率的
宏师姐 20:28:36
过渡态直接ts就可以了,可以和频率同时算。不需要再优化
宏师姐 20:29:13
出现虚频是很正常的,要把他们消去。只有一个的虚频才算是过渡态
宏师姐 20:31:26
不用,可以再优化,有时候如果结构有对称性可能会出现虚频。或者你上网上查怎么消虚频,然后把它消去,消去以后在优化。过渡态比较难找的
宏师姐 20:32:25
我消虚频的办法是找到虚频对应的坐标把它加在新的优化好的坐标上,然后重新优化。
宏师姐 20:33:31
只要有一个虚频都可以做irc,也就是你只要找到过渡态就可以做irc
宏师姐 20:43:44
很小的虚频也不行的,过渡态只能有一个虚频。过多的虚频是要消去的,那就表明你现在的结构不是过渡态,那么你在做IRC是没有意义的
小木虫上说
对于irc不收敛你可以加iop(1/13=5)试试
关于过渡态与反应物接近,还与是与产物接近,有一个它Hammond's Postulate,大意是:
如果在反应过程中有两个状态(比如说一个过渡态和一个反应中间体)相继出现,并且这两个状态的能量相差不大,那么这两个状态相互转变时的分子构型也不会有太大变化。
这个假定的一个有用推论就是,过渡态的分子结构会与在能量上和它相近的组分相似。如果一个反应是放热反应,那么反应物的能量高于产物的能量,但过渡态能量最高,在能量上过渡态就与反应物相近,根据Hammond's Postulate,它的分子结构也与反应物相近。这个过渡态就是前过渡态。
要是走构型就直接
# b3lyp/6-311g** IRC(MAXPOINT=400,CALCFC,stepsize=5,MAXCYCLE=600,forward) SCF(MAXCYCLE=400)
1.请问,在IRC计算中出现L508错误应该怎么办啊? 谢谢
scf cannot converge even using scf=qc,
there is no good enough solution for this error
but to use better initial guess for calculation.
e.g. use guess=read to read scf from similar molecule ( cation, anion, different spin states, neibor in perodic table, etc.); or change starting geometry
link下面加上这个geom=check guess=check
频率
在进行Gaussian计算时,经常会出现多个虚频个,每个都只有-30~-20左右,但是如果结构改动过大,则一不小心,虚频就会更大。消除虚频的主要方法是改变构型。其次在计算上还可以尝试:nosymm;加大循环次数;提高收敛度;iop(1/8=1)等。通常对于势能面很平,较小的虚频很难消除。
为了解决以上的这种现象,按如下的步骤一步步调整:
1). 在优化时采用Scf(tight)的选项,增加收敛的标准。再去计算频率。如果还有虚频,参见下一步。
2). 对称性的影响,很多情况下的虚频是由于分子本身的对称性造成的。这样,在优化时,如果必要,要将对称性降低,还有,输入文件有时是用内坐标。建议如果有虚频的话,将内坐标改成直角坐标优化。但是有时这一方法也并没有多大作用。
3). 如果上述方法还有虚频,看一下虚频,找到强度较大的,将在频率中产生的原子的振动坐标加到相应的输入文件中。这样,重新计算。直到虚频没有。比较好的做法是分子结构输入用直角坐标,将这些产生虚频的振动坐标直接与原始坐标相加。加的时候不一定要将坐标直接相加,可以选择权重如1/3相加等。
4). 实际上,如果分子柔性较大,很难找到最低点,这是电子结构计算的问题,这种情况下,需要动力学的 东西,用构象搜寻的办法解决。如:模拟退火,最陡下降法,淬火法等。将得到的能量最低的构象做一般的电子结构计算,这样,应当没有问题。
当出现两个以上虚频时,可以先看一下振动模式。如果多余的虚频,和想要的振动模式发生在不同原子上,可以先固定过渡态模式所对应的原子,然后做opt到minimal,一般可以把多余的虚频去掉,再放开做全优化。
opt(maxcyc=500)这么的
Opt=(QST2,maxcyc=500)
错误提醒:CConnectionGLOG::Parse_GLOG()
Gaussian error detected
Last line read = 218856
这个错误是你把东西考到你的D盘或者别的盘里就能打开了
gv有时吧U盘当作中文目录
you see??
如果不收敛时强制收敛,QC表强制的意思,CONVER=6表示收敛到小数点后六位,原来是八位
scf=(qc,conver=6)
用Gaussian进行溶剂计算
用SCRF关键词,可以指定它给出的系列溶剂(默认为H2O)可供选择的有19种溶剂,可以在SCRF关键词里面查到,也可以写成SCRF=N (N=1到19,分别对应N所代表的溶剂也可以用自己设定的溶剂但是要在输入它的介电常数和溶剂半径A0介电常数可以查化学手册,A0可以用Volume关键词进行计算计算方法可以选择PCM和Dipole两种模型e.g. # RHF/6-311++G(d,p) OPT=Vtight Freq SCRF(PCM,Solvent=4) Test 在甲醇溶剂中用PCM方法进行优化和频率计算
Frequence calculations must be run as a seperate job step for SCRF calculations.(Opt Freq does not do what might be expected)
Reference: Exploring chemistry with electronic structure methods Running SCRF calculation. so this example is not accurate.
错误提示: 我觉得是,类似的问题如下
ZZZY= -36.8108 XXYY= -3348.7631 XXZZ= -2487.6433 YYZZ= -1137.9718
XXYZ= 94.1140 YYXZ= -65.0780 ZZXY= -70.7703
N-N= 2.709998116592D+03 E-N=-1.489621355248D+04 KE= 3.981653343482D+03
Exact polarizability: 490.904 -43.677 431.547 42.754 -2.982 143.667
Approx polarizability: 521.672 -44.663 496.046 54.897 -8.847 176.906
D1PCM: PCM CHGder 1st derivatives, ID1Alg=0 FixD1E=F DoIter=F I1PDM=0.
D2PCM: PCM 2nd derivatives, FixD2E=F I1PDM=0.
D2PCM-2 allocation failure: iend,mxcore= 119167574 99361975
Error termination via Lnk1e in e:\g03w\l701.exe at Mon Jun 28 05:48:45 2004.
Job cpu time: 0 days 6 hours 5 minutes 37.0 seconds.
File lengths (MBytes): RWF= 943 Int= 0 D2E= 0 Chk= 3 Scr= 1
Sum of APT charges= 0.35270:电荷总和不为零.但Sum of Mulliken charges= 0.00000.此外错
误还在: D2PCM-2 allocation failure.但不知该怎样处理,还请大家帮忙看看.多谢!
会不会是内存不够?
"%mem=95MW"内存设置的是760MB,总共只有1G 的内存,就是在往大里设,也大不了多少了.不过很有
可能是内存的问题,因为我作了几个小分子的运算(在同一水平上),没出错.现在已把内存设到820MB(我
用的是XP 操作系统,本身就需要很大的空间),到明天看情况如何吧.多谢!
内存不足,我计算的分子比你的少10 来个原子,也必须用900M 内存呢
我在rb3lyp/3-21g*和CIS/sto-3g 下运算,一出错就是加内存条,逼得我重新买了一块主板,已经1.28G
内存了,大一点的基组还是不能用。
骄阳(250573296) 9:00:55
mxcore= 106368996 104443047
前面是你需要的最小空间,后面是你给的空间
骄阳(250573296) 9:01:15
应该是读写文件比较大,放不下了
%rwf=
分开写几个
试试看
%rwf=e:\1,2gb,e:\2,2gb,e:\3,2gb,-1(可以多建几个的.)
这个命令的意思是在e 盘下建立四个RWF 文件,前三个的容量大小均为2G,名字分别为
1.2.3。最后-1 表示e 盘下所剩的可用空间为第四个RWF 的空间。不过高斯对单个RWF 文件
的大小有限制,所以第四个RWF 的空间也应该不超过2G 才对。如果将e 改为F,则表示在
F 盘下建立一个RWF 文件。这样我们就可以在不同的分区下建立RWF 文件了。
加极化函数
这个使用程序自带的,自定义的是这样加的 (大致是这样的)
%chk=a.chk
%mem=800mb
%nproc=2
# opt(maxcyc=500) freq b3lyp/gen
0 1
……
C 0
6-31G
****
C 0
d 1
0.6
****
这是加一个指数为0.6的d极化函数。
呵呵,关于极化函数表,我用的是香港科技大学林镇阳先生的,如果需要,我可以发给你。老林是做金属有机非常好的,他说内地第一批公费出国的人员。对于有些加,有些不加大问题,一般说来,要反应的原子、也就是即将断裂或新生成键的原子必须加,其他可以不加。
关于一个问题:
一、请问大家下面的输入文件该怎么写啊?我刚学不久 希望那位高手能帮好好解释一下 谢谢
先用小基组,低精度算法优化一下,以得到较好的初始构型,再用高精度的计算接着算。如果前面的方法保留了chk文件,重新计算时需要使用 geom=allcheck 读入构型(就不必麻烦地写构型了), guess=read(读入初始波函数,可以加快第一步SCF收敛)。
1 # opt HF/sto-3g
2 # opt b3lyp/6-31g geom=check guesss=chk
先进行第一步计算,然后进行第二步计算,这之间不能改CHK文件的名字,路径也要一样,就OK了
另外选用什么样的基组,要根据你的体系来确定,呵呵
二、[求助]在Gaussian运行中出现Error termination via Lnk1e in d:\beifen\gw98-5.4\l609.exe.错误,用什么方法可以解决?其命令行是
#p Density=Current AIM=All b3lyp geom=connectivity gen pseudo=read
是scf不收敛,要用scf=qc
【分享】Gaussian计算中分子总能量各项的意义
在HFSCF方法计算中,体系采取固定核近似后,体系的哈密顿算符分为电子哈密顿和核哈密顿两部分,电子哈密顿分为三部分,电子动能,电子间斥能,及核电排斥,通过单电子近似,得到单电子的HF方程,自洽场的结果得到了电子的能量.核哈密顿分为两部分,核动能及排斥,由于核固定动能为零,只剩下一项,因此体系的能量就是电子能量加上核斥能,,这就是我们计算得到的分子总能量,因为分子在OK时仍在平衡位置振动,要加上零点校正.最后结果即例子中的:
Sum of electronic and zero-point Energies= -113.837130
在分子的热力学计算中,包括了零点能的输出,零点能是对分子的电子能量的矫正,计算了在0K温度下的分子振动能量.但我们计算的热力学数据并不总是在OK时,为了计算在较高温度下的能量,内能也要考虑到总能量中,它包括平动能,转动能,和振动能.注意在计算内能的时候,已经考虑了零点能.下面是一个计算实例
Temperature 298.15 Kelvin. Pressure 1.0000 Atm.
Zero-point correction= 0.029201
Thermal corection to Energy= 0.032054
Thermal correction to Enthalpy= 0.032999
Thermal correction to Gibbs Free Energy= 0.008244
Sum of electronic and zero-point Energies= -113.837130
Sum of electronic and thermal Energies= -113.834277
Sum of electronic and thermal Enthalpies= -113.833333
Sum of electronic and thermal Free Energies= -113.858087
后面四行的四个能量分别为E0, E, H, G. 计算为
E0=E(elec) + ZPE
E=E0 + E(vib) + E(rol) + E(transl)
H=E + RT
G=H - TS
上例是在298.15K和1.0Atm下计算的热力学状态函数,EO即我们的分子体系总能量,而在求298.15K下的内能E时,要加上平动能,转动能,和振动能,由统计热力学知,这三项处于激发能级,注意这里的ZPE实际上就是对应E(vib)在OK时的能量,但现在并非0K,有关能量随温度变化公式可参考统计热力著作,最后得到的E就是平常所指的内能U,这正是统计热力学中对内能的定义.对H,G状态函数,不再叙述.
由于本版无法上传公式,还请见谅,有关内容请参看相关著作
Gaussian中关于基组设置的简单介绍
基组有两种,一种是全电子基组,另一种是价电子基组。价电子基组对内层的电子用包含了相对论效应的赝势(缩写PP,也称为模型势, 有效核势,缩写ECP)进行近似。以铟原子为例,可以输入下面的命令来观察铟原子3-21G 基组的形式:
# 3-21G GFPrint
In
0,2
In
输出结果为:
3-21G (6D, 7F)
S 3 1.00 N=1 层,s 轨道
.1221454700E+05 .6124760000E-01
.1848913600E+04 .3676754000E+00
.4063683300E+03 .6901359000E+00
SP 3 1.00 N=2 层,s 和p 轨道
.5504422550E+03 -.1127094330E+00 .1523702990E+00
.1197743540E+03 .8344349530E-01 .6096507530E+00
.3866926950E+02 .9696880360E+00 .3970249500E+00
SP 3 1.00 N=3 层,s 和p 轨道
.4702931320E+02 -.2758954250E+00 -.1408484750E+00
.2249642350E+02 .5977348150E-01 .5290866940E+00
.6697116970E+01 .1082147540E+01 .6620681110E+00
D 3 1.00 N=3 层,d 轨道
.1021735600E+03 .1205559000E+00
.2839463200E+02 .4884976000E+00
.8924804500E+01 .5850190000E+00
SP 3 1.00 N=4 层,s 和p 轨道
.6572360380E+01 .4284830560E+00 .1091304620E-01
.2502157560E+01 -.4633643610E+00 .5036758870E+00
.9420245940E+00 -.8219679320E+00 .5581808650E+00
D 3 1.00 N=4 层,d 轨道
.4535363700E+01 .2508574000E+00
.1537148100E+01 .5693113000E+00
.4994922600E+00 .3840635000E+00
以上都是内层电子轨道,每个轨道用三个高斯型函数拟合,
也就是3-21G 中的3。
SP 2 1.00 N=5 层,s 和p 轨道
.1001221380E+01 -.4364171950E+00 -.2316333510E-01
.1659704190E+00 .1189893480E+01 -.9903308880E+00
这是价电子轨道,每个轨道用二个高斯型函数拟合,也就是3-21G 中的2。
SP 1 1.00 N=6 层,s 和p 轨道
.5433974090E-01 .1000000000E+01 .1000000000E+01
这是空轨道,每个轨道用一个高斯型函数拟合,也就是
3-21G 中的1。
可见3-21G 是全电子基组。再看看铟原子LanL2DZ 基组的结果:
LANL2DZ (5D, 7F)
S 2 1.00
.4915000000E+00 -.4241868100E+01
.3404000000E+00 .4478482600E+01
S 1 1.00
.7740000000E-01 .1000000000E+01
P 2 1.00
.9755000000E+00 -.1226473000E+00
.1550000000E+00 .1041757100E+01
P 1 1.00
.4740000000E-01 .1000000000E+01
这些都是价电子轨道。在ECP 上加上了双zeta(DZ)基组。所以LanL2DZ 是价电子基组。
在GAUSSIAN 中,CEP 也是很重要的价电子基组,并且98 版中对元素的适用范围扩大到了86号元素Rn。需要注意的是,超过了Ar 元素后,每一种元素的CEP-4G,CEP-31G,CEP-121G 是等价的(和加了极化*的CEP 还是有区别的)。SDD 适用于除了Fr 和Ra 以外的所有元素,但是结果的准确性并不太好。另外,在下面的两个网址,有几乎整个周期表元素的赝势下载,不过我想不出那些短寿命的超铀元素有什么用:
http://www.mapr.ucl.ac.be/ABINIT
http://www.theochem.uni-stuttgart.de
需要注意的是有些方法不需要输入基组,像各种半经验方法,G1, G2 等等。
收敛种类
自洽场收敛是指几何结构固定,从给定初始轨道猜测得到得到Hartree-Fock轨道,是与与轨道有关的(g03中的guess关键词),因为分子轨道是由原子轨道线性组合得到的,如何得到这样的组合系数使得轨道满足一定的条件小总的能量最低。从数学上讲就是用迭代法解一组非线性的方程组。
几何收敛是从给定一个分子的初始结构得到一个驻点(能量对坐标的一阶导数为零),驻点的性质(局域最小点或者过渡态或者其它)通过二阶导数(频率分析)来确定
(g03中的opt和freq关键词)。比如对于优化局域最小点,从数学上讲就是对于一个多维函数,通过某种优化方法,找到它的最小值。
出现自旋污染怎么解决
Annihilation of the first spin contaminant:
S**2 before annihilation 1.0749, after 0.7526
Convergence failure -- run terminated.
一般来说,自旋污染超过了10%的计算结果都需要重新审视的,尤其是DFT计算的结果。。
解决的方法很简单,Stable测试以及Stable优化,在优化的得到的波函数的基础上再优化结构。
Stable测试:# HF/6-31+g* stable ,注意只要里边有一个负值就表示不稳定,结果会明确提示你稳定与否的
Stable优化:# HF/6-31+g* stable =opt 要保留CHK 文件
再优化结构:一定要从刚才得到的CHK文件中读取波函数,然后:# HF/6-31+g* opt freq 等等的。。
在一个机器上加几组计算任务
windows下高斯多步计算,不知道该怎么弄? 就是先优化完一个结构之后,自动开始下一个优化(和第一个不是同一个文件),谢谢
有没有输入文件的例子啊?
%chk=a.chk
%mem=800mb
# hf/3-21g opt
optA
0 1
A结构坐标
(空格)
--link--
%chk=b.chk
%mem=800mb
# hf/3-21g opt
optB
0 1
B结构坐标
用bcf文件就可以啊
如
!
!user created batch file list
!start=1
!
e:\cal\CH4-td-opt.gjf , e:\cal\CH4-td-opt.out
e:\cal\C2H6-631g-opt.gjf , e:\cal\C2H6-631g-opt.out
【原创】几个处理输入输出文件的shell 脚本。
Linux 下的shell 脚本是一种批处理语言,可以把单个的命令组合起来,又能够进行判断循环等流程控制。
当面对成百上千个的输入输出文件需要操作时候,除了shell,你还能选择什么呢?这里列出几个常用的操作,并给出相应的解析,希望能对大家有所帮助。
2-5楼
1 把当前目录下所有的gjf 文件用g03 计算
2 修改gjf 的开头
3 把out文件的能量提取出来,存储到get_energy.log 中,同时生成以能量值为文件名的out文件,便于排序筛选
4 把out文件的最后一个坐标做成gjf 文件,并赋予新的关键词
10楼 使用方法
all_g03.sh
#!/bin/bash
for inf in *.gjf
do
outf=`echo ${inf}|tr"gjf" "out"`
g03 < ${inf} >${outf}
done
该文件可以把当前目录下所有的gjf文件用g03 计算,文件名后缀自动改为out。
解析
#!/bin/bash
在shell 脚本中,以#开头的行是注释,但在文件最开始的以#! 开头的行指定解析这个脚本所用的程序。此处是bash,还可以是csh,python, tcl等若干。
for inf in *.gjf
do
… …
Done
循环,变量为inf,变量取值为当前目录下的所有*.gjf 文件。
outf=`echo ${inf}|tr"gjf" "out"`
${inf}: shell中变量取值时候需要在前面加$, 当变量名为多个字符时候最好用{}
echo ${inf}: 显示inf 的值,此处在每次循环时候为某一个*.gjf 的文件名
echo ${inf}|tr "gjf""out" : 把inf 替换为out,为同windows 下的g03兼容,用out做后缀
outf=`echo ${inf}|tr "gjf""out"` :给outf 赋值
g03 < ${inf} >${outf}
用g03 计算,从inf 输入,outf 输出
change_keyword.sh
#!/bin/bash
dir=ckout
mkdir ${dir}
for i in *.gjf
do
echo "#b3lyp/6-31g opt" >> ${dir}/${i}
echo "" >> ${dir}/${i}
echo "${i}" >> ${dir}/${i}
echo "" >> ${dir}/${i}
echo "0 1" >> ${dir}/${i}
sed '1,5d' ${i} >> ${dir}/${i}
echo "" >> ${dir}/${i}
done
此脚本可以批量修改gjf 的开头,输出文件在一个新的文件夹中,文件名不变。
解析
dir=ckout
mkdir ${dir}
创建一个叫ckout 的目录,用于存放新生成的文件。
echo "#b3lyp/6-31g opt" > ${dir}/${i}
echo "" >> ${dir}/${i}
echo "${i}" >> ${dir}/${i}
echo "" >> ${dir}/${i}
echo "0 1" >> ${dir}/${i}
新gjf 文件的开头部分,可以根据需要修改。
echo"" >> ${dir}/${i}:写入一个空行
sed '1,5d' ${i} >> ${dir}/${i}
把旧的gjf 文件的坐标部分写入新文件,
'1,5d' 删除(d) 文件的1-5行,5 可以根据需要修改。
echo "" >> ${dir}/${i}
在文件末尾写入新的空行
get_energy.sh
#!/bin/bash
dir=get_energy
mkdir ${dir}
for i in *.out
do
en=`more ${i} | grep 'Energy'| tail -n 1 | cut -c 13-20`
cp -- ${i} ${dir}/${en}_${i}
echo "${i} ${en}" >> get_energy.log
done
此脚本可以把out文件的能量提取出来,存储到get_energy.log 中,同时生成以能量值为文件名的out文件,便于排序筛选。
解析
en=`more ${i} | grep 'Energy'| tail -n 1 | cut -c 13-20`
获取能量值。
grep'Energy': 对不同的文件,能量的表达不同,am1用”Energy”,hf 用E(RHF),可以根据实际选择。
Tail –n 1: 对于opt 任务,此处可以选择最后一个能量值
cut -c 13-20:截取能量值,13-20个字符,未必就是这么多,可以通过试用得到。注意前面不要有空格和减号。
cp ${i} ${dir}/${en}_${i}
复制文件到新的目录,并命名为
能量_原文件名
echo "${i} ${en}" >> get_energy.log
统计能量到新的文件。
out2gjf.sh
#!/bin/bash
dir=out2gjf
mkdir ${dir}
for i in *.out
do
fil=`echo ${i}|tr "out" "gjf"`
n=`more ${i}|cat -n|grep Number|tail -n 1|cut -c 1-8`
((n=n+2))
echo "#hf/3-21g opt" > ${dir}/${fil}
echo "" >> ${dir}/${fil}
echo ${i} >> ${dir}/${fil}
echo "" >> ${dir}/${fil}
echo "0 1" >> ${dir}/${fil}
ido=0
while((${ido} < 30))
do
((m=n+ido))
more -- ${i} |head -n${m}|tail -n 1|cut -c 14-99 >> ${dir}/${fil}
((ido=ido+1))
done
echo "" >> ${dir}/${fil}
done
此脚本可以把out文件的最后一个坐标做成gjf 文件,并赋予新的关键词。
解析
fil=`echo ${i}|tr "out" "gjf"`
给变量fil 赋值,输入文件的后缀名改变为gjf
n=`more ${i}|cat -n|grep Number|tail -n 1|cut -c 1-8`
寻找最后一个坐标所在的位置,n为行号。
ido=0
while((${ido} < 30))
do
((m=n+ido))
more -- ${i} |head-n ${m}|tail -n 1|cut -c 14-99 >> ${dir}/${fil}
((ido=ido+1))
done
把分子坐标写入新的gjf 文件
这里30 为分子的原子数,使用时根据体系原子数修改
说一说这些脚本的使用方法。
以第一个为例
首先给那个all_g03 加上可执行权限
chmod u+x all_g03.sh
然后把它放到一个PATH 包含的路径下面,进入存放gjf 的目录,输入all_g03.sh &,
或是把它放到那些gjf 里面,输入./all_g03.sh,这时那个脚本会依次计算这个目录下的所有gjf 文件。后面加& 实在后台执行,这时可以输入exit,关闭终端,回去睡觉好了 。
。
这些脚本都很简单,可以根据自己的实际情况修改或扩展。比如输入文件后缀为com,程序为g98 等,都可以修改。还可以加一句,在计算结束之后关机。
**** Warning!!: The largest alpha MO coefficient is 0.11062683D+02
分子轨道的系数超过了100
Gradient too large for Newton-Raphson use scaled descent instead
是用了最速下降法取代了牛顿法进行局部优化
沙漠之鹰<mr.cobra@qq.com> 9:24:28
局部优化的方法通常有爬坡法、最速下降法和牛顿法,其中爬坡法适用于任何情况,最速下降法要求势能面的一阶导数不变号,牛顿法则要求二阶导数也不变号,在后面的帖子中会陆续介绍。收敛速度最快的算法适应的条件最苛刻。在guassian中采用一种称为Berny搜索的算法,实际上是对这三种算法的综合运用
NBO
1.NBO(Natural Bond Orbital),自然键轨道分析程序,在量化软件Jaguar,Q-Chem,PQS,NWChem,GAMESS-US(含PC GAMESS)和Gaussian中都包含了此程序或接口。目前是5.0版。
2.Gaussian-03 users should therefore include IOp(5/48=10000)
3.#p B3LYP/Gen pseudo=read Pop=NBORead pop=reg
.........
.........
C O H 0
6-31G(d)
****
Zn 0
LANL2DZ
****
Zn 0
LANL2DZ
$nbo bndidx $end
4. NPA相对于传统的Mulliken集居数分析,具有更高的数值稳定性,能更好地描述有较高离子特征化合物的电子分布,克服了Mulliken集居数分析方法的弱点:会产生没有物理意义的负集居数值;Mulliken 集居数随所选基组不同差异很大,尤其是当基组扩大到更高精度时会失去意义;对于具有明显离子特征的化合物而言,Mulliken集居往往给出不合理的电荷分布图。而且自然集居数分析基于正交归一化基组横跨整个轨道空间,本质上是波函数,而不是筛选的特殊轨道基,因而不论基组怎样扩大,其占据数都能收敛于一较稳定值。
5. PBC:周期性边界条件,算高分子,晶体等的,但在用NBO的时候可能不好用。
6.NBO输出文件:
Bond types; Hybrid descriptors



BD for 2-center bond,(二中心键) BD是成键轨道,包括sigma和pi键
CR for 1-center core pair,
LP for 1-center valence lone pair, (孤对电子)
RY* for 1-center Rydberg(里德堡), 我们一般计算的激发态称为Low-lying Excited States,也就是由原子价电子构成的分子电子激发态。更高的电子态是Rydberg态。从头算法和其他半经验方法对空轨道,特别是较高空轨道的计算结果不好,因而得到的Rydberg态的结果缺乏明确的物理意义。
BD* for 2-center antibond,
Bond orders


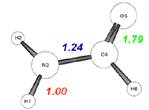
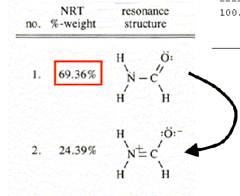
T:total c:covalent i:ionic(这是讨论键的极性,是共价键为主还是离子键为主)
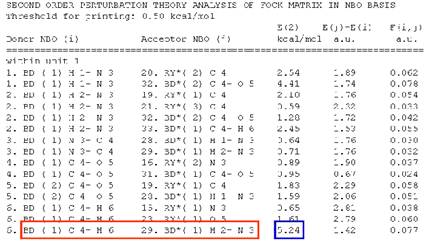
Charge transfer
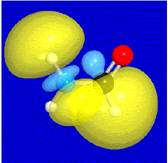

Resonance weights