第一章 有机化学实验的基本知识
1.1 实验室安全规则和实验室守则
有机实验室与其它化学实验室相比可能是更危险更易发生事故的地方,因为有机化学实验所用药品一般都是易燃易爆、有毒或强腐蚀性的,使用不当就会引起着火、爆炸、烧伤、烫伤、冻伤或中毒等事故。另外,碎裂的玻璃器皿、煤气或电器设备使用不当也会引起事故,所以在实验中要严格执行操作规程,遵守有机实验室安全规则和实验室守则,进行有机实验时必须遵守如下规则:
1. 进入实验室前,充分预习好实验内容,开始实验前做好各项准备工作,要写出预习报告。
2. 进入实验室后应保持安静,实验过程中不能擅自离开岗位。
3. 实验前应检查仪器设备是否存在破损、漏气、漏电等不安全因素,实验中应注意观察现象,如发现异常,应立即中断实验。
4. 处理有毒或有强刺激性气体时要在通风橱中进行。
5. 减压操作或处理爆炸性物质及强腐蚀性物质等可能发生危险的实验,应使用防护眼镜、面罩、手套等防护设备。
6. 公用仪器及药品用后立即归还原处。节约用水、电、煤气及药品。药品及实验后的产品不能随意丢弃以免酿成事故。
7. 严禁在实验室内吃食物、吸烟,实验后要洗手。
1.2 事故的预防及急救措施
1. 防止火灾及急救措施
防火的基本原则是使火源远离有机试剂,并尽可能减少有机物挥发到空气中。如果已经着火,很小面积的着火可用湿抹布、石棉布或用砂土盖住即可,大面积着火应用灭火器扑灭。
2. 使用易燃易爆气体,如氢气、甲烷、乙炔等要打开室内排风装置且严禁明火及防止一切火星发生。
3. 常压操作时整套装置应有一处与大气相通,切不可形成封闭体系,以免因体系内压太大而使反应物冲出或玻璃仪器炸裂。减压蒸馏时,蒸馏瓶和接收瓶应用梨形或圆形、茄形烧瓶,以免平底瓶因其受压不均而炸裂。
4. 处理强酸、强碱、强氧化剂及其它有剧毒的药品时要带橡皮手套;不要用敞口容器存放有毒的药品。
5. 处理危险性化合物要严格遵守操作规程。对于久置后会产生易爆物质的过氧化物,应预先处理后方可使用
6. 使用电器前应检查是否漏电或其它不正常现象,不可用湿手接触电插头。不慎触电时,应立即切断电源,必要时进行人工呼吸。
7. 实验室应备有急救箱,箱内常备如下药品和工具:医用70%乙醇、2%醋酸、1%硼酸、3~5%碳酸氢钠、红药水、甘油、烫伤膏、消炎粉、硼酸软膏、创可贴;医用剪刀、镊子、洗眼杯、消毒棉、纱布、胶布、绷带、棉签等,发生事故时如伤情较轻按以下方法处理:
(1)割伤:清洗伤口后涂上红药水或创可贴,用绷带扎住。
(2)烫伤:不要用水冲洗,可在烫伤处涂上烫伤膏。
(3)试剂烧伤
酸:量少时立即用大量水冲洗,再用3~5%的碳酸氢钠水溶液洗,最后再用水洗,然后涂上硼酸软膏(或氧化锌软膏);量大时,必须用洁净的柔软物质迅速将酸擦去,再按照酸量少时的办法处理。
碱:立即用大量水冲洗后,用2%的醋酸水溶液洗,最后再水洗。若为钠灼伤,用镊子将钠移走后,再按照碱灼伤时相同办法处理。
溴:立即用大量水冲洗,再用酒精擦至创面处无溴液时为止,然后涂上甘油。
(4)试剂溅入眼内:任何试剂溅入眼内,都要先用大量水冲洗,直至眼睑内没有为止;如溅入眼中的是玻璃,则应仔细地用镊子小心地把玻璃夹出,然后再水洗,严重的经初步处理后要送医院。
(5)中毒:毒品溅入口中若无咽下者立即吐出,再用大量水冲洗口腔;如已吞下,应根据毒物性质给以解毒剂。
腐蚀性毒物:对于强酸或强碱,应先饮大量水,然后分别服用氢氧化铝、鸡蛋白、醋或酸果汁,最后用牛奶灌住,不要吃呕吐剂。
刺激性神经性毒物:先饮牛奶或鸡蛋白,再食用约30g硫酸镁溶于一杯水中的溶液催吐,在进行初步处理后,伤情较重者要送医院。
1.3 实验预习、实验记录和实验报告
1.3.1实验预习
实验预习认真与否,常常是实验做得是否成功、收获大小的关键之一。应当做到不了解一个实验的目的和操作原理以前不进行实验。因此,要求每个学生都应准备一个实验记录本,在充分预习好实验内容的基础上,写出预习报告,预习报告应包括如下内容:(1)实验题目(2)实验目的(3)实验原理,反应方程式(包括主要副反应)(4)主要试剂、主、副产物的物理常数(5)实验装置图(6)实验操作、现象和结果记录表。(7)对反应操作时间上的合理安排。
1.3.2实验记录
进行实验时不仅要做到操作认真、观察仔细,而且要积极思考,并将观察到的现象(如变色、沉淀、气泡等)和测得的各种数据(如熔程、沸程、加热温度等)如实地及时记录于记录本中,不允许做回忆笔记。合成反应要记录产物的外观、物理状态和产量。记录要做到简明扼要,字迹清楚。实验结束后,将记录本交指导教师签字,并上交产物。
1.3.3实验报告
写实验报告是完成实验工作不可缺少的环节。通过写实验报告、分析实验现象、归纳整理实验结果,可以把从实验中得到的感性认识上升到理性认识,这对今后的工作和科研是一种很好的训练,因此必须认真对待。
1. 合成实验报告的内容:(1)实验目的(2)实验原理(合成原理、分离原理)
(3)主要试剂及产物的物理常数(4)仪器及药品规格、用量(5)实验装置图(6)实验步骤及反应现象(7)产率计算(8)回答问题及讨论。
2. 下面是乙醚的制备实验报告,可供参考。
乙醚的制备
一、实验目的
1. 了解酸催化下乙醇分子间脱水制备乙醚的原理。
2. 掌握低沸点化合物的蒸馏操作。
二、实验原理
1. 合成原理
低级伯醇在酸性脱水剂催化下,共热生成单醚。
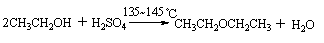 主反应:
主反应:
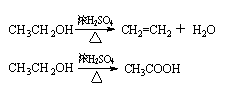 副反应:
副反应:
2. 粗产物分离纯化原理
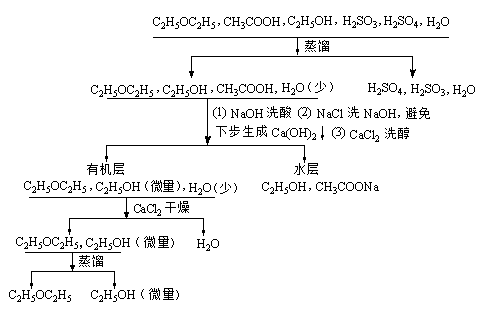
三、物理常数

 名称 分子量 性状 密度 熔点 沸点
名称 分子量 性状 密度 熔点 沸点
 (℃) (℃)
(℃) (℃)
乙醇 46.07 无色液体 0.7898 -117.3 78.5 ∞ ∞ ∞
乙醚 74.12 无色液体 0.7138 -116.2 34.51 微溶 ∞ ∞
 乙酸 60.05 无色液体 1.0492 16.6 117.9 ∞ ∞ ∞
乙酸 60.05 无色液体 1.0492 16.6 117.9 ∞ ∞ ∞
四、仪器与药品
1. 仪器及规格
三颈瓶:100mL;滴液漏斗:60mL;分液漏斗:125mL;直型冷凝管:20mL;圆底烧瓶:50mL;接引管。
2. 药品规格及用量
所用药品皆为分析纯,用量如下:
乙醇(95%):37.5mL(29.6g,0.61mol); 浓硫酸:12.5mL;
饱和氯化钠溶液:7.5mL; 饱和氯化钙溶液:7.5mL;
5%氢氧化钠:7.5mL
五、实验装置图
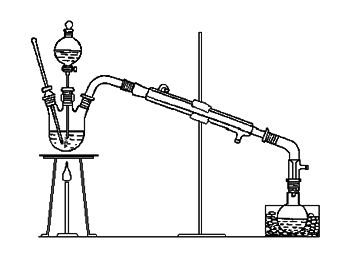
图1–5 乙醚制备装置
六、实验步骤及现象

实 验 步 骤 现 象 备 注
 1. 用量筒取12.5mL 95%乙醇于三颈瓶中 溶液无色
1. 用量筒取12.5mL 95%乙醇于三颈瓶中 溶液无色
2. 边摇边缓慢加入12.5mL浓 H2SO4。 瓶内有少许白烟,瓶壁很热
3. 取25mL95%乙醇于滴液漏斗中,按图 温度迅速上升,溶液迅速变
装好仪器。加入1~2粒沸石,石棉网直火 黄,瓶内有大量白烟(SO3)
加热,使温度迅速升到140℃。
4. 由滴液漏斗慢慢滴加乙醇使与蒸馏液 有馏份蒸出,温度保持
流出速度相等,大约每秒1滴,保持温 不变(141℃)
度在135~145℃之间。
5. 乙醇加完,继续加热至160℃,停火。 温度上升,不再有馏份
6. 馏出液转入125mL分液漏斗中,加 振荡后静置分层,上层澄
7.5mL 5%NaOH溶液洗一次 清。分出下层,弃去
用7.5mL饱和NaCl洗涤有机层 上层、下层都澄清
用7.5mL饱和CaCl2洗涤有机层 上层澄清、下层浑浊
7. 分出下层,将醚层从分液漏斗上口
倒入100mL锥形瓶中,加约2g无水 溶液无色澄清
氯化钙,塞上塞子,并不时振荡,放
置20min
8. 将产物过滤到50mL圆底烧瓶中, 温度计读数34℃时,有
用热水浴(50~60℃)加热蒸馏, 馏份馏出,至37℃时温
收集33~38℃馏份。 度不再上升。
9. 产物外观、质量 无色澄清液,8.2g

七、产率计算
实际产量
 产率 = ×100%
产率 = ×100%
理论产量
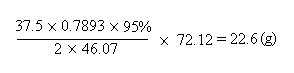
理论产量 =
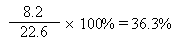
产率 =
八、回答问题及讨论
1. 为什么不采用回流装置?
答:因为该实验为可逆反应,采用蒸出产物(醚和水)可推移平衡,有利正反应进行。
2. 温度计和滴液漏斗为什么要插入液面下?
答:因为要测量反应液的温度,故必须把温度计插入液面以下。另外,反应时容器内压力较大,滴液漏斗颈如不插入液面以下,会使液体(乙醇)滴不下来。因此建议以后用恒压滴液漏斗。
3. 为什么在开始时乙醇—浓硫酸混合液温度超过乙醇沸点而乙醇却蒸不出来?
答:因为此时乙醇和浓硫酸已形成酸式硫酸酯。
讨论:
1. 本实验控制好温度很关键,如低于130℃则不宜成醚,高于160℃易生成烯。
2. 控制滴加乙醇的量等于蒸出乙醚的量最为合适,否则乙醇滴得过快反应来不及进行就将被蒸出,乙醇滴得过慢则延长反应时间。
3. 乙醚沸点低,故接收器放在冷水浴中以减少挥发损失,其支管接上橡皮管通入水槽或窗外,是防止乙醚蒸气与空气形成爆炸极限,以保证安全。
4. 本实验开始时将H2SO4往乙醇里加时,由于未充分冷却且加入速度太快,溶液颜色变黄(甚至变黑)说明有副反应发生。在粗产品洗涤时分层处受外界光线影响看不清,加之放液快了些,当发现时已有少量醚随水层放出。为回收这部分醚,将分出的水层放入另一分液漏斗中静止,但很长时间都没发现醚层出现,这可能是由于醚量少而溶解在水层中,另外也可能挥发掉了。由于上述补救办法没有成功,故计算产率偏低。今后在使用分液漏斗时,放液要慢,观察要仔细。
第二章 有 机 实 验
实验一 苯甲酸合成
实验目的
1. 了解甲苯氧化法制备苯甲酸的原理,
2. 复习回流、减压过滤和重结晶基本操作。
实验原理
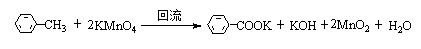
烷基苯在高锰酸钾的氧化下生成苯甲酸。
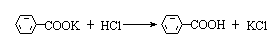
药品
甲苯:3.5mL(6.1g,0.033mol) 高锰酸钾:10.4g(0.053mol)
浓盐酸;亚硫酸氢钠
或者:
甲苯:1.0mL(0.9g,0.0094mol) 高锰酸钾:2.0g(0.0106mol)
浓盐酸;亚硫酸氢钠
实验步骤
在50mL园底烧瓶中加入2.0g高锰酸钾和28mL水,摇匀,尽量使之溶解。加2粒沸石,安装冷凝管,小火加热至沸。从冷凝管上端滴加1.0mL甲苯,整个加料过程约需10min,继续煮沸,直到回流液不再有明显油珠(约50min)。如果反应混合物显较深的紫色,加入少量亚硫酸氢钠,振摇使紫色褪去后,再趁热过滤,用少量热水(约5~10mL)洗涤残渣。将滤液放在冷水浴中冷却,用浓盐酸酸化直至溶液呈强酸性,苯甲酸全部沉淀析出为止。抽滤,用少量冷水洗涤,尽量抽干。把苯甲酸在表面皿上摊开晾干并称重。粗产品可用热水重结晶。纯苯甲酸为无色针状晶体,熔点为122.4℃。
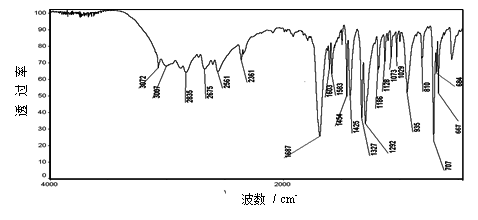
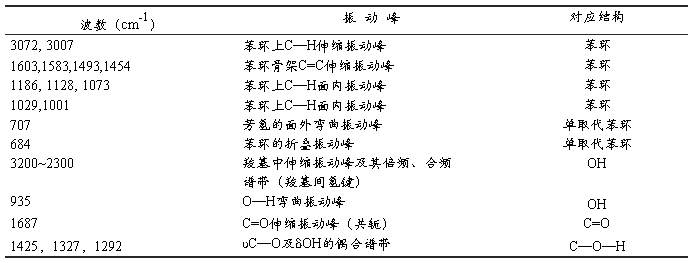
图3–17 苯甲酸红外光谱图
表3–2 苯甲酸的红外光谱解析
思考题
1. 反应结束后,反应混合物呈紫色时为什么要加入少量亚硫酸氢钠?
2. 如何判断酸化过程中已呈强酸性?
3.用什么方法精制苯甲酸?
实验二 从茶叶中提取咖啡因
实验目的
1. 了解从茶叶中提取咖啡因的原理。
2. 巩固回流、蒸馏基本操作并学习沙浴的使用方法。
实验原理
 茶叶中含有多种生物碱,其中以咖啡碱(又称咖啡因)为主,约占1~5%,另外还含有11~12%的丹宁酸(又称鞣酸)及0.6%的色素纤维蛋白质等。咖啡碱是弱碱性化合物,易溶于氯仿(12.5%)、水(2%)和乙醇(2%)等,在苯中的溶解度为1%(热苯为5%)。丹宁酸易溶于水和乙醇,但不溶于苯。
茶叶中含有多种生物碱,其中以咖啡碱(又称咖啡因)为主,约占1~5%,另外还含有11~12%的丹宁酸(又称鞣酸)及0.6%的色素纤维蛋白质等。咖啡碱是弱碱性化合物,易溶于氯仿(12.5%)、水(2%)和乙醇(2%)等,在苯中的溶解度为1%(热苯为5%)。丹宁酸易溶于水和乙醇,但不溶于苯。
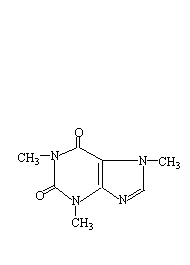
咖啡因是杂环化合物嘌呤的衍生物,它的化学名称是1,3,7-三甲基-2,6-二氧嘌呤,其结构式如下:
含结晶水的咖啡因是无色针状结晶,味苦,能溶于水、乙醇、氯仿等。在100℃时即失去结晶水,并开始升华,120℃时升华相当显著,至178℃时升华很快。无水咖啡因的熔点为235.5℃。为了提取茶叶中的咖啡因,往往利用适当的溶剂(乙醇、氯仿、苯等)在提取器中连续提取,然后蒸去溶剂,即得粗咖啡因。粗咖啡因还含有其它一些生物碱和杂质,利用升华可进一步提纯。
咖啡因具有刺激心脏,兴奋大脑神经和利尿等作用,因此可作为中枢神经兴奋剂,也是复方阿司匹林(A.P.C.)等药物的组分之一。
药品(半微量法)
茶叶末:约1g 95%乙醇:10mL 生石灰:0.5g
实验步骤
常量法:实验装置参见图2–22。称取10g茶叶末放入脂肪提取器的滤纸套筒(1)中,在圆底烧瓶内加入80mL 95%乙醇,用水浴加热,连续提取2~3h(2)后待冷凝液刚刚虹吸下去时,立即停止加热。然后改成蒸馏装置。回收提取液中的大部分乙醇,再把残液倾入蒸发皿中,拌入3~4g生石灰粉(3),在蒸气浴上蒸干,最后将蒸发皿移至煤气灯上加热使水分全部除去。冷却后擦去沾在边上的粉末,以免在升华时污染产物。取一只合适的玻璃漏斗,罩在隔以刺有许多小孔的滤纸的蒸发皿上,用沙浴小心加热升华(4)。当纸上出现白色毛状结晶时,暂停加热,冷至100℃左右,揭开漏斗和滤纸,仔细地把附在纸上及器皿周围的咖啡因用小刀刮下,残渣经拌和后再用较大的火加热片刻,使升华完全。合并两次收集的咖啡因,测定熔点。若产品颜色不纯时,可用少量热水重结晶提纯(或放入微量升华管中再次升华)。
半微量法:在50mL圆底烧瓶中加入10mL 95%乙醇,瓶口用一带勾接头连接,勾上挂一多孔玻板提取器,内置约1g茶叶末,接头上接水冷凝管,见图2–23。小火加热至沸,连续提取40min,停止加热,改成蒸馏装置,进行蒸馏。待瓶内液体剩2~3mL时,停止蒸馏。将残液倒入蒸发皿中,并加入0.5g氧化钙粉末,拌成糊状,用小火加热至干(须边加热边搅拌)。取一只合适的玻璃漏斗,罩在隔以刺有许多小孔的滤纸的蒸发皿上,用沙浴小心加热升华。约20min升华结束,揭开漏斗和滤纸,仔细地把附在纸上的咖啡因用小刀刮下。
注释
(1)滤纸套大小既要紧贴器壁,又能方便取放,其高度不可超过虹吸管;滤纸包茶叶末时应严紧,防止漏出堵塞虹吸管。纸套上面折成凹形,以保证回流液均匀浸润被萃取物。
(2)若提取液颜色很淡时,即可停止提取。
(3)生石灰起吸水和中和作用,以除去部分杂质。
(4)在萃取回流充分的情况下,升华操作的好坏是本实验成败的关键。在升华过程中,始终都需用小火间接加热。温度太高会使滤纸炭化变黑,并把一些有色物烘出来,使产品不纯。第二次升华时,火亦不能太大,否则会使被烘物大量冒烟,导致产物损失。