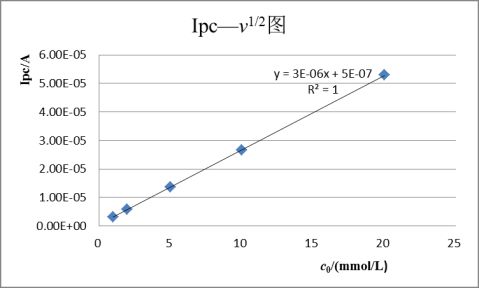
循环伏安法
实验结果的记录与处理:
第二篇:物化实验报告_凝固点降低法测定摩尔质量[1]
凝固点降低法测定摩尔质量
1.1实验目的
1. 用凝固点降低法测定萘的摩尔质量。
2. 通过实验掌握溶液凝固点的测量技术,并加深对稀溶液艺术性之的理解。
1.2 实验原理
稀溶液具有依数性,凝固点降低是依数性的一种表现,固体溶剂与溶液成平衡时的温度称为溶液的凝固点。依数性即指定溶剂的种类和数量后,这些性质只取决于所含溶质的分子数目,而与溶质的本性无关。它与溶液质量摩尔浓度的关系为:
其中,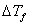 为凝固点降低值,
为凝固点降低值,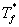 、
、 分别为纯溶剂、溶液的凝固点,
分别为纯溶剂、溶液的凝固点,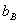 为溶液质量摩尔浓度,
为溶液质量摩尔浓度,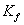 为凝固点降低常数,它只与所用溶剂的特性有关。如果稀溶液是由质量为
为凝固点降低常数,它只与所用溶剂的特性有关。如果稀溶液是由质量为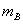 的溶质溶于质量为
的溶质溶于质量为 的溶剂中而构成,则上式可写为:
的溶剂中而构成,则上式可写为:
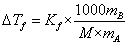
即
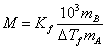 (*)
(*)
式中: ——溶剂的凝固点降低常数(单位为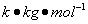 );
);
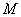 ——溶质的摩尔质量(单位为
——溶质的摩尔质量(单位为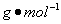 )。
)。
如果已知溶液的值,则可通过实验测出溶液的凝固点降低值,利用上式即可求出溶质的摩尔质量。
常用溶剂的值见下表。
表1 常用溶剂的值
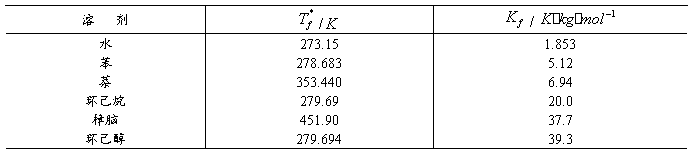
实验中,要测量溶剂和溶液的凝固点之差。对于纯溶剂如图1(a)所示,将溶剂逐渐降低至过冷(由于新相形成需要一定的能量,故结晶并不析出),温度降低至一定值时出现结晶,当晶体生成时,放出的热量使体系温度回升,而后温度保持相对恒定。对于纯溶剂来说,在一定压力下,凝固点是固定不变的,直到全部液体凝固成固体后才会下降。相对恒定的温度即为凝固点。
对于溶液来说,除温度外还有溶液浓度的影响。当溶液温度回升后,由于不断析出溶剂晶体,所以溶液的浓度逐渐增大,凝固点会逐渐降低。因此,凝固点不是一个恒定的值。如把回升的最高点温度作为凝固点,这时由于已有溶剂晶体析出,所以溶液浓度已不是起始浓度,而大于起始浓度,这时的凝固点不是原浓度溶液的凝固点。要精确测量,应测出步冷曲线,按下一页图1(b)所示方法,外推至校正。
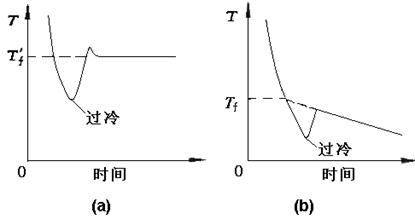
图1 溶剂和溶液的步冷曲线
2 实验操作
2.1 实验药品、仪器型号及测试装置示意图
1.仪器
TSW-3型 精密数字温度计(-12-12℃,0.002℃)
CJ-2型 磁力搅拌器
HS-4型 精密恒温浴槽(可制冷,-20-100℃)
冷阱 大试管 移液管(25ml)分析天平(公用) 秒表
2.药品
环己烷(分析纯,密度0.778g/ml),萘(分析纯,摩尔质量:128.17)
3.测试装置示意图(见右图)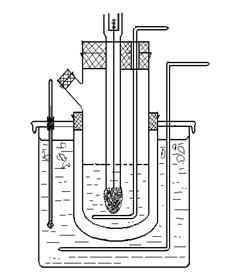
图2 凝固点降低法测定摩尔质量实验装置示意图
2.2 实验条件
表2 实验条件

2.3 实验操作步骤及方法要点
1.仪器装置
仪器装置如上一页图2所示,将恒温槽温度调至2℃左右,通入冷阱。在室温下,用移液管移取25ml环己烷,加入大试管。调整数字温度计的测温探头,使探头顶端处于液体的中下部。调整磁力搅拌器转速旋钮至适当旋转速度,保持恒定。
2.溶剂凝固点的测定
观察数字温度计的变化,此时温度逐渐降低,当温度降低到最低点之后,温度开始回升,说明此时晶体已经在析出。直到升至最高,在一段时间内恒定不变。此时温度即为溶剂的凝固点,记下温度值。
取出大试管,不要使溶剂溅到橡皮塞上,用手捂住试管下部片刻或用手抚试,用手温将晶体全部融化(注意不要使温度升高过多,避免以后实验的降温时间过长)。
将大试管放回冷阱,重复上述操作。如此再重复数次,直到取得三个偏差不超过 0.005℃的值。取其平均值作为溶剂环己烷的凝固点。
0.005℃的值。取其平均值作为溶剂环己烷的凝固点。
3.溶液凝固点的测定
取出大试管,在管中加入0.15g左右(准确到0.0002g)的萘,注意不要粘于管壁上(可先将萘压成片)。拿掉磁力搅拌器上的冷阱,将大试管直接放在磁力搅拌器上搅拌至全部融解,然后按装置图将仪器装好。观察数字温度计,当温度降至6℃时打开秒表,半分钟记录一次温度值,当温度降至最低开始回升后每15秒记录一个值,到最高点,将最高点温度记录下来(用此点估算萘的摩尔质量)。此后再恢复到半分钟记录一个值,记录6~7个点,实验结束。
环己烷溶液倒入回收甁。
3 结果与讨论
3.1 原始实验数据
1) 原始实验相关数据记录
表3 原始实验相关数据记录表

2) 原始实验测量数据
(a)溶剂凝固点测定原始数据
表4 溶剂凝固点测定原始数据记录表
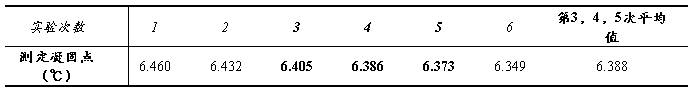
在实验中,一共进行六次凝固点测定,取其中差距最小的三次,取平均值后作为溶剂的凝固点,即溶剂凝固点Tf*=6.388℃。
(b)溶液凝固点测量原始数据
表5溶液凝固点测量原始数据记录表
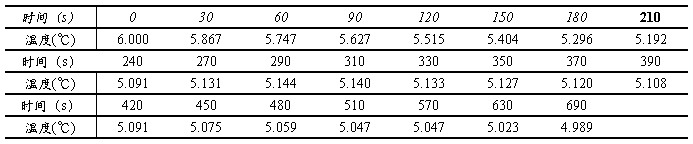
3.2计算的数据、结果
a) 利用冷却曲线计算溶液凝固点Tf
利用Origin Pro软件将表5中的数据用作图法表示出来,可得到实验中溶液的冷却曲线。结果如下图3所示:

图3 测定所得的溶液步冷曲线。测定体系由0.1525g萘与25.0mL环己烷组成,当系统温度降至6.000℃时开始记录,横坐标为时间,纵坐标为所测得的溶液温度,从而得到溶液的步冷曲线。
为得到准确的溶液凝固点,将溶液凝固后温度回升又下降的部分做反向延长线,与开始的温度下降线相交,分别选取线性程度较好的有效数据区进行线性拟合,所得到两条直线的交点即为溶液的凝固点Tf。结果如下图4所示:
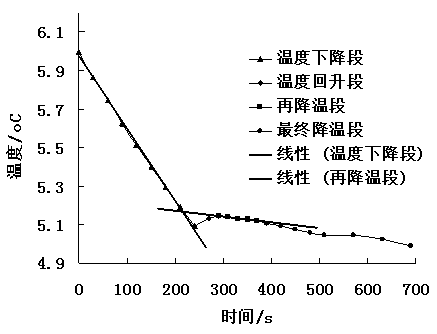
图4 步冷曲线有效数据区线性拟合结果。步冷曲线即图3中曲线,取开始温度下降段和再降温段进行线性拟合,所得直线的交点即为所求溶液的凝固点。
根据图4所得到的结果,两条曲线的线性拟合结果分别为:
y = -0.0038x + 5.979(R2=0.9985),y = -0.0003x + 5.2334(R2=0.9928)。
联立两个方程,求解两直线交点的坐标,可得溶液凝固点的温度值为:Tf = 5.169℃。
综上,用步冷曲线法得到溶液凝固点为5.169℃。
b) 根据凝固点变化计算萘的摩尔质量
利用前述凝固点测定结果,可根据公式(*),即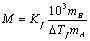 计算萘的摩尔质量。
计算萘的摩尔质量。
由已知: ΔTf= Tf*- Tf= 6.388℃ - 5.169℃ = 1.219℃ = 1.219K
mB= 0.1525g,mA = ρ x V = 0.778×25.0 = 19.45g,Kf = 20.0 K·kg·mol-1
将以上数据代入前述计算公式,得
萘的摩尔质量为MB= 20.0 × 1000 × 0.1525 / (1.219 × 19.45) = 128.6 g/mol
3.3讨论分析
1) 测定数据处理结果的误差分析及其与文献值的比较
(a)测定溶质(环己烷)凝固点的结果与文献值的比较
由文献值(见表3)可知,环己烷凝固点为6.3°C,而本次实验中的最终结果为6.388°C。将实验值与文献值进行比较可知:实验结果与文献值的相对误差ε = (6.388-6.3/6.388)×100%=1.378%。这一误差相对而言是比较大的,说明我们的测量和文献值有较大的偏差。
(b)测定溶质(环己烷)凝固点的测量误差分析
测定溶质凝固点时,对于三组数据(4中第3,4,5组的数据)进行误差分析可得:
平均误差: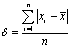 ,可得δ = 0.0113
,可得δ = 0.0113
标准误差: ,可得σ = 0.0161
,可得σ = 0.0161
因此测量结果精确度可用标准误差表示为:Tf*=6.388±0.0161 (℃)
(c)测定溶质(环己烷)凝固点过程中产生误差的可能原因及分析
对溶质环己烷的凝固点测量中,产生误差的可能原因如下:
首先,我们在测定过程中的每一次测定的条件可能不同,从而导致测定结果的误差较大。例如,测定时虽然我们十分注意仪器的装配,但如果大试管的管壁不慎碰到了冷阱,很有可能导致所测定的温度不准确。同时,磁力搅拌器的转速不均匀或者中途发生变化可能导致溶质结晶过程中各部分的热传导率等物理化学性质不均一,从而导致测量误差。另一方面,综合各次测定的结果,我们发现每一次测量误差的绝对值和符号总是使测量结果永远朝一个方向偏,由此推断在测定过程中存在系统误差。对于系统误差的具体分析请参见下文中对异常现象的分析和讨论。
(d)测定萘摩尔质量的结果与文献值的比较
由文献值(见表3)可知,萘的摩尔质量为128.17 g·mol-1,而本次实验中根据测定数据计算出的最终结果为128.6 g·mol-1。将实验值与文献值进行比较可知:实验结果的相对误差ε = (128.6-128.17/128.6)×100%=0.334%。误差相对而言比较小,说明我们得到的实验结果值与文献值相当接近。因此,我们在实验所采取的测定萘的摩尔质量的方法相对而言还是比较准确的。
2) 对实验中异常现象的分析和讨论
(a) 对溶剂(环己烷)凝固点测定中异常系统误差的分析
综合各次对溶剂(环己烷)凝固点的测定结果,我们发现每一次测量误差的绝对值和符号总是使测量结果永远朝一个方向偏,由此推断在测定过程中存在系统误差。其分析如下:
一方面,进行前两次测定时我们调整过磁力搅拌器的转速,可能使测量结果不准确,但在随后的四次测定中系统的配置没有过任何改变,因此最有可能的解释是搅拌器本身转速不稳定。另一方面,由于在随后的溶液凝固点测定过程中没有出现温度测定的异常情况,所以可以排除数字温度计出现故障的可能。此外,还有一种可能是我们所取的环己烷含有杂质。但是由于其他组没有出现相同的问题,所以可能是在向大试管转移液体的过程中引入杂质。最后,我们认为比较关键的原因是:我们组的实验位置距离恒温槽较近,因此在测量过程中,冷阱中循环水的温度很可能始终偏低,从而导致我们的测定结果始终向一个方向偏移。
(b) 对溶剂(环己烷)步冷曲线中稳定温度的分析
测定溶剂凝固点的过程中,实验的最后阶段应能够测得一个比较稳定温度值。但是在达到稳定之前,我们发现温度计的显示数值在很小的范围内(约0.002℃)会有短暂时间的波动,而且这种波动是无规律的。形成这个现象的可能原因有:当过冷液体刚刚开始结晶时,溶液处于两相共存的状态,但是由于搅拌不可能完全均匀,因此使得体系各个部分的温度和物理性质在不均匀的变化(特别是热传导性质和密度)。因此温度探头所接触部分的液体温度有可能产生无规则变化。
3) 对仪器装置、操作步骤、实验方法的改进意见和实验体会
完成全部实验和后期数据处理后,我深切地感受到这次实验利用我们所掌握的物化知识,从实验的角度对我们进行了一次充分的训练。最后所取得的实验结果是次要的,而我们所掌握的物化实验方法和实验过程的总结才是关键。结合实验具体过程和感受,我对本项试验有如下几点建议:
(a) 数据记录过程:在本次实验过程中,数据是人为判读和记录的,因此很可能会引入偶然误差,可以考虑利用一台同步记录仪进行温度数值的记录。
(b) 恒温槽的功能:在本次试验中由于恒温槽的限定,使得溶液降温的速度基本是恒定的,如果可能的话可以考虑使用一种降温速度可调整的恒温槽,在不需要记录温度的开始阶段快速降温,在数据记录段则正常降温。
(c) 装置的设计:试验中大试管的管壁较容易碰到冷阱,如能够在大试管和冷阱之间增加一个密封圈(可采用橡胶质地),就可以使得大试管的定位更加稳定,对增强实验结果的稳定性和准确性有益。
(d) 实验的重复型检验:一次实验并不能得到令人信服的结论,测量对象,测量条件的变化和仪器带来的系统误差都会影响到实验结果的重复性。因此,如果要验证贝克曼法的准确程度和适用范围,我们还需要进行进一步的实验,比如更换其他的溶质,或者对步冷曲线进行多次测量检验等。
4 结论
本次实验通过凝固点降低法(贝克曼法)测定了萘的摩尔质量,测定结果汇总如下:
环己烷纯溶剂凝固点: Tf*= 6.388℃。
萘的环己烷溶液凝固点:Tf = 5.169℃.
萘的摩尔质量: MB = 128.6 g/mol
本实验的结果与文献值相比的误差很小,证实了利用此方法测定摩尔质量的可行性与准确性。同时,本实验也验证了稀溶液依数性理论的正确性和实用性。在一定条件下,我们可以把较稀的溶液当作理想溶液来处理,利用其依数性测定有关的物理化学性质。而由此得到的结果和真实值的误差较小,在精确度要求不是非常高的情况上可以满足测定的要求。
5 参考文献
[1]张连庆等.步冷曲线法-对凝固点降低测定摩尔质量的改进.大学化学.北京:大学化学编辑部,2006.第21卷.第二期.54~56.
[2]清华大学化学系物理化学实验编写组.物理化学实验.北京:清华大学出版社,1991.45~53.
[3]朱文涛.物理化学(上册). 北京:清华大学出版社,1995.190~192.
[4]王军民 薛芳渝 刘芸 物理化学.北京:清华大学出版社 1993.
[5]孟尔熹, 实验误差与数据处理, 上海:上海科学技术出版社,1988.
6 附录
思考题:
1)实验中所配溶液浓度,太浓或太稀都会使实验结果产生较大的误差,为什么?
答:在本次实验中,溶液的浓度应使得测定过程中有部分溶质凝固析出,但还有一部分剩余溶液,且其浓度应当保持在一定的值。因此,如果溶液太稀,可能在析出的时候会完全析出,从而使得测量的凝固点温度不准确。另一方面,溶液浓度太小会使得凝固点的变化量太小,从而使实验中的误差对结果的影响很大,造成结果不准确。而如果溶液太浓,首先就会使得溶液的性质不符合稀溶液的依数性原理,使得计算公式不适用于所测定的体系,造成较大的实验误差。
2)测凝固点时,纯溶剂温度回升后能有一相对恒定阶段,而溶液则没有,为什么?
答:由分析纯级别的环己烷所构成的溶剂在凝固时所生成的晶体纯度很高,性质均以稳定,晶格排列较为规则整齐。因此,在测量仪器可探测得范围内,纯溶剂的温度回升后会遵循理想晶体凝固的性质而具有固定的凝固点(此时属于固相、液相共存状态),即实验中的温度相对恒定阶段。而溶液由于在凝固过程中不断发生组成变化, 其凝固温度就不断降低, 直到另一组分(溶质)也从溶液中饱和析出,因此没有温度相对恒定阶段。
3)为什么会产生过冷现象?
答:在凝固过程中,新生的固相在生成时由于其颗粒直径很小,比表面能较高,所以体系很难自发地向能量较高的方向进行。此时即便温度降低到相变点(凝固点)以下,相变过程也无法发生,从而导致体系的温度低于凝固点,即过冷现象(此体系成为过冷液体)。在此状况下,若体系的能量升至很高或收到外界干扰(比如投入大小适宜的晶核),液体即会迅速凝固为固体,发生相变过程。
4)原理中计算公式的导出作了哪些近似处理,如何判断本实验中这些假设的合理性?
答:在计算公式的推导过程从 出发,进行了一下几项近似处理:
出发,进行了一下几项近似处理:
a) 测定溶液符合稀溶液依数性,为理想稀薄溶液(溶质分子在溶液中的活度近似等于1)。
b) 纯溶剂的摩尔溶化焓不随温度T变化。
c) Tf*·Tf = (Tf)2
d) 析出的第一粒固体是纯固体
实验中溶剂的浓度很小,所以析出的固体可近似认为是纯固体。溶液可以近似认为是理想稀薄溶液。因为DTf较小,其变化而导致摩尔熔化热的变化可以忽略,也因此可以近似认为Tf*·Tf = (Tf)2。最终实验结果与理论值较吻合,说明推导过程中的这些近似是合理的。