原子吸收分光光度法测定水样中钠离子含量
班级:09化工-3班 姓名:肖普 学号:0910370315
一、 实验目的
1.掌握原子吸收分光光度法测定水样中钠离子含量的分析方法;
2.学习正确使用原子吸收分光光度计。
二、 实验原理
1、 原子吸收光谱的产生
任何元素的原子都是由原子核和绕核运动的电子组成,原子核外电子按其能量的高低分层分布而形成不同的能级。因此,一个原子核可以具有多种能级状态。能量最低的能级状态称为基态能级(E0=0),其余能级称为激发态能级,而能最低的激发态则称为第一激发态。正常情况下,原子处于基态,核外电子在各自能量最低的轨道上运动。如果将一定外界能量如光能提供给该基态原子,当外界光能量E恰好等于该基态原子中基态和某一较高能级之间的能级差△E时,该原子将吸收这一特征波长的光,外层电子由基态跃迁到相应的激发态,而产生原子吸收光谱。电子跃迁到较高能级以后处于激发态,但激发态电子是不稳定的,大约经过10-8秒以后,激发态电子将返回基态或其它较低能级,并将电子跃迁时所吸收的能量以光的形式释放出去,这个过程称原子发射光谱。可见原子吸收光谱过程吸收辐射能量,而原子发射光谱过程则释放辐射能量。核外电子从基态跃迁至第一激发态所吸收的谱线称为共振吸收线,简称共振线。电子从第一激发态返回基态时所发射的谱线称为第一共振发射线。由于基态与第一激发态之间的能级差最小,电子跃迁几率最大,故共振吸收线最易产生。对多数元素来讲,它是所有吸收线中最灵敏的,在原子吸收光谱分析中通常以共振线为吸收线。
2、 原子吸收光谱分析原理
由于原子蒸气最容易吸收的光是它的特征谱线频率的光,分析原理是将待测元素在高温下原子化产生原子蒸气,然后用光源(一般为同元素物质做成的空心阴极灯)辐射出的待测元素的特征光谱通过样品的蒸汽中被待测元素的基态原子吸收后,测定发射光被减弱的程度,进而求得样品中待测元素的含量,由于吸收的
化工专业实验
过程符合Lamber?Beer定律:
I??I0,?e(?k?L) ?lgI0,??0.434K?L?A I0
? A???b?c ? A?c
因此通过测定未知浓度元素的吸光度与已知的标准溶液吸光度进行对比便可计算出待测元素浓度。
3、 仪器构成
原子吸收分光光度计基本结构如图1所示:
图1 原子吸收分光光度计结构

数据处理系统
光源:发射待测元素的锐线光谱,一般采用空心阴极灯作为光源。
原子化器:产生高温将样品液中的元素原子化。
分光系统(单色器):将空心阴极灯阴极材料的杂质发出的谱线、惰性气体发出
的谱线以及分析线的邻近线等与共振吸收线分开。
检测器:检测光的强度,产生电信号。
数据处理系统:计算出元素的浓度。
三、 操作步骤
1、 配置标准液
在6个100mL容量瓶中分别加入100mg/L钠标准液0、0.2、0.4、0.6、0.8、1.0mL,各加入2%的稀硝酸0.3mL,用蒸馏水定容至刻度线并摇匀。

1
化工专业实验
2、 水样消噪
采用硝酸消解法处理待测水样,稀释待测。
3、 开机
打开主机电源,检查排液管水封是否正常。
4、 软件操作
打开电脑,进入BRAIC操作界面。
(1) 点击编辑新方法,选择火焰原子吸收,然后选择要测量的元素,记住编号,点击确定。
(2) 分析条件选择:选择待测元素灯位置,设定阻尼常数为2,工作曲线参数中选择标准空白。
(3) 点击文件?新建?火焰原子吸收? 确定。
(4) 分析任务设计:点击 元素 ?灯位置?选择方法?选择要测的元素?确定?样品表?添加的待测样品?确定。
(5) 仪器控制:点击 波长设置?灯位置调节,通过点击“上、下”来调节,使得主光束最大?自动增益,使主光束达到100%左右。
5、 点火
打开空气压缩机,调节空气出口压力到100MPa左右,打开乙炔气阀,调节压力至0.05~0.1MPa,顺时针旋转主机乙炔流量至最小,按点火开关,听到“啪”的声音后,调节流量至1~1.5/min。
6、 绘制标准曲线及确定样品浓度
测量之前先调零两到三次直至稳定,将毛细管以此浸入到标准溶液,点击“读数”,之后点击并显示“标准曲线”。清洗毛细管,测定水样,点击“数据”显示水样浓度。
7、 关机
(1) 先关闭乙炔钢瓶总阀,待管道中乙炔全部燃尽后在关闭乙炔流量计。
(2) 关闭空压机阀门。
(3) 关闭主机电源
(4) 退出程序,关闭电脑。
2
化工专业实验
四、 数据处理
1、数据记录

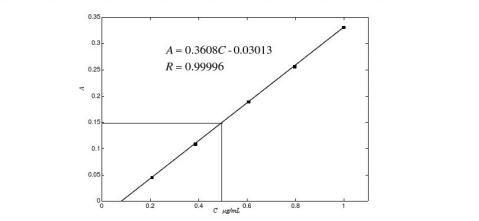
2、绘制标准曲线
计算待测液浓度:
0.1484?0.03013C??0.4948 ?g/mL 0.3608
3
化工专业实验
4
第二篇:液相色谱实验报告
仪 器 分 析 实 验 报 告

实 习 名 称: 液相色谱分析实验
学 院: 化学工程学院
专 业: 化学工程与工艺
班 级: 化工班
姓 名: 学 号
指 导 教 师:
日 期:
一、实验目的
1、了解液相色谱的构造、原理及操作技术;
2、了解并熟悉液相色谱仪的流程及操作方法。
二.实验原理
液相色谱由泵、进样器、色谱柱、检测器、记录仪等几部分组成。
高效液相色谱分析的流程。由泵将储液瓶中的溶剂吸入色谱系统,然后输出,经流量与压力测量之后,导入进样器。被测物由进样器注入,并随流动相通过色谱柱,在柱上进行分离后进入检测器,检测信号由数据处理设备采集与处理,并记录色谱图。废液流入废液瓶。遇到复杂的混合物分离(极性范围比较宽)还可用梯度控制器作梯度洗脱。这和气相色谱的程序升温类似,不同的是气相色谱改变温度,而HPLC改变的是流动相极性,使样品各组分在最佳条件下得以分离。
高效液相色谱的分离过程。同其他色谱过程一样,HPLC也是溶质在固定相和流动相之间进行的一种连续多次交换过程。它借溶质在两相间分配系数、亲和力、吸附力或分子大小不同而引起的排阻作用的差别使不同溶质得以分离。
开始样品加在柱头上,假设样品中含有3个组分,A、B和C,随流动相一起进入色谱柱,开始在固定相和流动相之间进行分配。分配系数小的组分A不易被固定相阻留,较早地流出色谱柱。分配系数大的组分C 在固定相上滞留时间长,较晚流出色谱柱。组分B的分配系数介于A,C之间,第二个流出色谱柱。若一个含有多个组分的混合物进入系统,则混合物中各组分按其在两相间分配系数的不同先后流出色谱柱,达到分离之目的。
不同组分在色谱过程中的分离情况,首先取决于各组分在两相间的分配系数、吸附能力、亲和力等是否有差异,这是热力学平衡问题,也是分离的首要条件。其次,当不同组分在色谱柱中运动时,谱带随柱长展宽,分离情况与两相之间的扩散系数、固定相粒度的大小、柱的填充情况以及流动相的流速等有关。所以分离最终效果则是热力学与动力学两方面的综合效益。
三.仪器及试剂
仪器:液相色谱仪(大连依利特)、微量注射器
试剂;乙腈和0.1%磷酸水溶液1:1混合液、样品(氨基布洛芬)
四.操作流程或步骤
1、准备(老师已备好)
(1) 准备所需的流动相,用合适的滤膜过滤,超声脱气;
(2) 根据待检样品的需要选择并安装好合适的色谱柱;
(3) 配置样品和标准溶液(也可在平衡系统时配制),用合适的滤膜过滤;
(4) 检查仪器各部件的电源线、数据线和输液管道是否连接正常;
(5) 开机:接通电源,依次开启不间断电源、泵、检测器,和检测器自检后,打开打印机、电脑显示器、主机,最后打开色谱工作站。
(6) 用样品液润洗注射器,润洗干净后,取20µL标准液,注入贮液器,观察电脑上峰的形成。
(7) 用润洗液润洗注射器,润洗干净后,取20µL样品也,注入贮液器,观察电脑上峰的形成,一次方法测量多组。
(8) 实验完成后,清理实验仪器,关闭相应仪器,最后关闭电源。
2、参数设定
(1)波长设定:在检测器显示初始屏幕时,按[func]键,用数字键输入波长值,按[enter]键确认。按[CE]键退出到初始屏幕,也可以选择LC-solution 软件方便地设定;
(2)流速设定:在泵显示初始屏幕时,按[func]键,用数字键输入所需的流速(柱在线时流速一般不超过1mL/min),按[enter]键确认。按[CE]键退出,也可以选择LC-solution 软件设定;
(3)流动相比例设定:在A泵显示初始屏幕时,按[func]键,用数字键输入流动相B的浓度百分数,按[enter]键确认。按[CE]键退出,也可在软件中设定。
3、更换流动相并排气泡
将A/B管路的吸滤器放入装有准备好的流动相的储液瓶中,逆时针转动A/B泵的排液阀180o,打开排液阀,按A/B泵的[purge]键,pump指示灯亮,泵大约以9.9mL/min的流速冲洗,3min(可设定)后自动停止,将排液阀顺时针转到底,关闭排液阀,如管路中有气泡,则重复以上操作直至气泡排尽。
4、平衡系统
(1) 按 泵的[pump]键,亮pump指示灯。用检验方法规定的 流动相冲洗系统,一般最少需6倍柱体积的流动相, 观测泵控制屏幕上的压力值, 压力波动应不超过1MPa。
(2) 观察基线变化。如果冲洗至基线漂移<0.01mV/min,噪声为<0.001mV时,可 认为系统已达到平衡系统,可以进样。
(3) 进样:将样品用滤膜过滤,用10uL玻璃注射器将样品液体从进样阀进(注意,进样时,尽量不要让针头接触进样孔的壁),进好样后,进行测试,观察工作电脑桌面上液相曲线变化,测试完后停止进样,用电脑进行数据分析。
5、关机 实验完毕后,关闭仪器和电脑。
五.实验参数
见附页
六.实验结论
1.已知液相关数据的计算
1)理论塔板数的计算
如图取第3组、第8组得
峰高的平均值
峰面积平均值A=(1825.31+1758.64)/2=1791.975 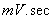
如图取第3组数据,
保留时间: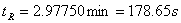

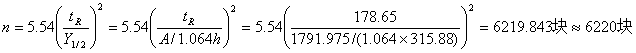
2)分离度的计算
取数据第2组,第3组数据
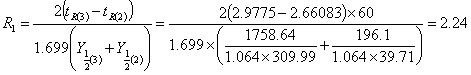
取第3组,第4组数据
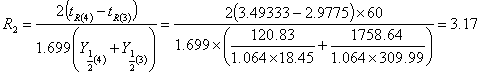
取:R=1.5作为相邻两峰完全分离的标准。即可得到如下结论:
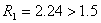 ,可认为完全分离。
,可认为完全分离。
 ,可认为完全分离。
,可认为完全分离。
2.未知液相关数据的计算
取第12组、16组、20组数据
峰高平均值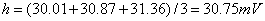
峰面积平均值A=(170.84+174.42+176.54)/3=173.93
1)理论塔板数的计算
同上代入第16组数据,如下
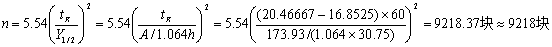
2)分离度的计算
取第15、16组数据:
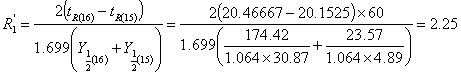
取数据第16、17组数据

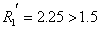 ,可认为完全分离。
,可认为完全分离。
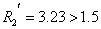 ,可认为完全分离。
,可认为完全分离。
七.结论
标准液峰分别是出现在2.97750min的第3个峰、出现在9.34500min的第8个峰;三次样品峰分别是出现在15.49583min的第12个峰、出现在20.46667min的第16个峰和出现在26.50883min的第20个峰。
由于实验组分比较简单,只要在实验过程中,严格控制且定量进样,就能得出准确的实验结果。若采用归一化法或内标法,虽然能得到准确更高的实验结果,但由于实验操作及计算均较为复杂,实验及数据处理的时间长,效率相对不高,故实验中采用外标法作为定量的方法。
最佳色谱条件的选择是本实验准确度的关键。进样针进样时确保进样针中没有残留气泡,另外,在进不同浓度或不同的物质前因先用甲醇溶液润洗干净,以免相互影响。
八.注意事项
(1) 流动相必须用HPLC级的试剂,使用前过滤除去其中的颗粒性杂质和其他 物质 。
(2) 使用缓冲溶液时,做完样品后应立即用去离子水冲洗管路及柱子,然 后用 甲醇(或甲醇水溶液)冲洗,以充分洗去离子。对于柱塞杆外部,做完样品后也必须用去离子水冲洗20ml左右。
(3) 每次做完样品后应该用溶解样品的溶剂清洗进样器。
(4) 气泡会致使压力不稳,重现性差,所以在使用过程中要尽量避免产生气泡。
(5) 如果进液管内不进液体时,要使用注射器吸液:通常在输液前要进行流动 相的清洗。
(6) 更换流动相时应该先将吸滤头部分放入烧杯中边振动边靖洗,然后插入新 的流动相中。
九.问题讨论
影响柱效能的主要因素有:填料、柱压、温度、流速。于是在实验过程中要注意仪器在运行中不能进入气体,否则会导致压力不稳定,影响柱效。
色谱柱标明了流体的流动方向,不要改变其流动方向,否则会降低色谱柱的性能。
为什么不能用纯乙腈作为流动相?因为这样会使单向阀粘住而导致泵不进液。