审稿意见答复
《色谱》编辑部:
谢谢审稿专家的宝贵意见!我们已经按照这些意见认真修改了文章。下面是对意见的具体答复,请审阅。
刘虎威,龙巍然
2012.04.10
1. 摘要请扩充至中文250-300字,英文200字以上。
答复1:之前的中、英文摘要都较为简略了,现已按要求做了扩充。
2. 图表中的文字、图表的注释,请全部用英文表示。
答复2:图表中的文字、图表的注释,已全部改为英文表示
3. 表4中的加标量是多少?并请补充加标回收率的RSD。
答复3:因为表4本身内容较多无法再增加内容,为了补充加标回收率的RSD,增加了一个表分别为:表5 市售六种饮料中食品添加剂含量测定结果。
4. 为了文中图片更加清晰准确,请提供您的论文中图的数据或原始谱图文件. 答复4:附上论文的原始谱图和图1的数据
5.附上图文摘要:
80810
6
5
47
13
11
12914
milliabsorbance units/mAU6016 Food Additives1517164032012
023568911
t/min
第二篇:审稿意见
意见如下:
1) 介绍中因说明镎这种元素的应用背景和产生的过程。
2) 融合极限通过混合自由能曲线的最小值来判断来进行判断,但是仅仅选用了三个点,既
镎的含量分别为0、50%、100%,选用的点有点少。
3) 计算融合能时候,没有给出所有位置的能量、键强数据,计算初始模型的构建没有说明
清楚,此外,反应物和生成物的物相(固体、液体,气体)对计算结果产生较大的影响,每个反应的计算过程中,选择什么样的状态没有给出说明,对计算结果的评估将造成一定的影响。
4) 计算过程中,不能仅仅考虑取代阳离子的位置,NP的位置也有两种。
5) 在一个反应过程中采用CASTEP和DMOL3相结合方法进行计算获得的能量,是否有其
它的例子来验证这种方法的可行性。此外,带电体系用CASTEP计算的主要问题是什么?
镎与铀酰根的融合:量子力学评估
摘要:
镎会融合到腐蚀的UO2变化的相中,实验表明层间带有正电阳离子的铀酰比层间不带阳离子的更容易融合镎,这种融合机制尚未明确。用密度泛函理论来比较镎融合到黄硅钾铀矿中的过程。电荷平衡机制的考虑包括:1)H+的增加;2)层间取代;3)层内取代。
源(镎)的选择,参与反应的阳离子的相影响了最终的计算结果及融合能。层间取代机制时,采用氧化氮(ΔEoxide= 2.4 eV),硅酸盐(ΔEsilicate= 1.2 eV)。源Np和释放阳离子采用典型水复合物,模拟水复合物交换的时候分别采用了团簇模型和周期性结构模型。对于H附加机制,由融入氧化物相(ΔEincorporation= 0.79 eV)的比融入水复合物相(ΔEincorporation= 0.66)更不容易。层内替换机制被用来预测Np融入到黄硅甲铀矿的极限,融入能大约为0.86eV,在300度是最大的融合量为585ppm。
1介绍:
锕系污染物影响环境和人类健康。锕系元素通过核废料包与地下水接触、铀矿床、核反应堆事故等可能被释放到环境中。环境中锕系元素的释放流动性取决于很多因素,但其中一个重要的过程是锕系元素融合到新的相中,如所使用的核燃料腐蚀产品。将锕系元素掺入矿物后形成的结构将大大限制其流动性。在氧化条件下,二氧化铀由一般的共生蚀变铀从铀硅酸盐/氢氧化物变成更可溶性磷酸盐阶或者碳酸盐,取决于具体的化学环境。镎和铀分子的相似之处,其化学性质也很相近,因此,镎容易融合到铀的不同的相中。
铀酰的不同相中可以融入不同量的NP,分析NP -掺杂的方法包括透射电子显微镜和同步技术,电子能量损失谱(EELS)和X -射线吸收光谱(XAS)。550 ppm的镎融合[三氧化铀· 0.8H2O]中。后来,同步XAS的数据显示,几乎没有纳入镎,结果导致这些矛盾的解释是多余的,因此,后来的研究,继续使用电子能量损失谱测量来量化成铀阶段的NP 融合。
进行一系列的实验,结果都被解释为是由于电荷平衡,加上替换机制,但是,在以前研究并没有提供关于原子尺度的替代机制的信息。通过在原子尺度和电子尺度模拟,了解NP –键合的环境变化,提供电荷平衡耦合取代机制的一种直观的认识。在这项研究中,采用量子力学计算,比较了各种机制的Np+5基态的融合量。
为什么要研究镎这种元素?镎的应用背景和产生背景??
2 黄硅钾铀矿:发生和结构:
一种天然铀(VI)硅酸盐,二氧化铀实验室腐蚀研究的中间产物。铀硅酸盐空间群为
P21 / m,铀五角双锥和硅氧四面体,其中的U(VI)与两氧组成UO22+分子,五个氧原子形成一个五边形,阳离子四面体的硅氧和OH -形成SiO3OH。形成的K /钠boltwoodite固溶。为了简化计算, boltwoodite夹层是由水和K组成(图1)。
计算方法:
3.1计算方法:
CASTEP,平面波基组,周期性边界条件,超软赝势描述心电子与价电子的相互作用,GGA(PBE)用来近似电子交换和相关性。此外,自旋极化近似考虑两个未成对的5F电子 (Np5+)电子,Cutoff为800, K-point为0.07 ? - 1。总能量收敛性为1.0.10 -5 eV /原子,几何收敛为2.10 -5eV /原子。
3.2能量定义
定量结果(焓,融合能)必须加以比较,结合实验结果来验证计算准确性。实验上,焓或形成能是固体测量获得的。但是,量子力学计算获得的能量不是形成能,根据不同的计算方法,是晶格能(使用经验力场)或零价气相元素的反应能(如CASTEP计算)。图2流程图给出了三种化学反应的描述,从铀的形成(一)自然零价,(二)气体零价元素,(三)离子,分别对应(一)形成能,(二)CASTEP能源,以及(三)晶格能。
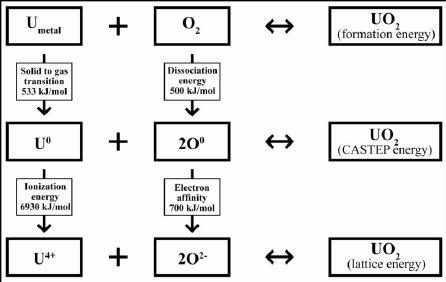
形成能由UO2和反应物(U和气态氧)能量差计算获得,其中气态氧气的氧分子为计算 在10 × 10 × 10一个盒子。
对铀的晶格能也可以计算,主要是在CASTEP中加入电离能(U0变成U4+的)和电子亲和力(O0变成O2-)。对电离能和电子亲和的值可采用高斯程序中量子力学计算或从实验测量。 Elattice = ECASTEP(UO2) – [(ECASTEP(U0) + IE)+2*(ECASTEP(O0) + EA)]
融合能的计算
融合能为NP取代U的反应过程的能量的变化。
K2(UO2)2(SiO3OH)2(H2O)3 + ?Np2O5 + ?O2 ? K2(UO2)(NpO2)(SiO3OH)2(H2O)3 + UO3 源是NP2O5,水复合物是UO3,生成物与反应物之间的能量差即为融合能,计算可以采用CASTEP计算获得的能量,也可以是形成能或是晶格能,只要每种化合物采用的标准一致即可。采用形成能或是晶格能同时提供了源与sink的相的信息。
融合能由依赖于融入机制,如H+增加或是层间取代,取代离子的位置以及取代反应发生时离子的相状态。比较了不同电荷平衡机制下的融入机制及离子取代位置;比较了H+融入机制下氧化物及水复合物的区别,比较不同层间的替代机制下氧化物和硅酸盐相的情况。
融合能高低的判断?应该是低的好吧?
3.3 评估NP的融入极限
实验是测量不到NP融入铀酰的能量的,实验上能测量到的是NP的融入量,如果能计算出NP的融入量对实验有很大的帮助。
如何从理论上来评估融入极限没太明白??从混合自由能曲线的最小值来判断。(11页)
4结果
4.1 取代机制1:Np5+ + H+ ? U6+
计算了四中H的取代位置:1)硅氧四面体与铀多面体间的O上;2)硅氧四面体顶部的O上;3)层间水分子形成水合氢;4)镎酰的氧上。(最稳定位置)
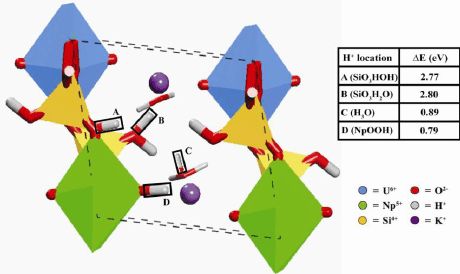
K2(UO2)2(SiO3OH)2(H2O)3 + ?Np2O5 + ?H2O ?HK2(UO2)(NpO2)(SiO3OH)2(H2O)3 + UO3
ΔEincorporation = 0.79 eV
其它位置的融合量、键强分别是多少?1.84与1.88不太一致,H的引入是否影响了所有的NP-O的键强??键长的这种解释是否合理呢?
(每个反应里面,反应物与生成物都是什么相?计算的初始模型都是如何构建的??能量定义是否前后一致,如NP2O5应该是固体吧? H2O呢?(液态)?如果不一致,是否进行转换了?
根据键价的计算,铀酰离子周围的键基本是满的,酰氧很少涉及其它附加的键,而镎酰的键强(键价)低于铀酰离子,键长较短(1.84),因此,镎酰的氧可以参与其它的成键。 而一个镎酰多面体氧附近镎酰多面体的配体。此外,H离子键合在镎酰氧上可能导致氧气是overbonded, NP - O键长的延伸,使中间层与H离子发生二次成键,足以让这个耦合取代发生。计算获得的OH - -Np5+的键长2.10,而其他镎酰的NP - O键的距离是1.88。在0.22 ?的延伸就是为了弥补O2 –的overbonding。
4.2取代机制II: Np5+ + Ca(Mg)2+ ? U6+ + K+
Ca(Mg)2+中间层中有两种可能的取代位置(K+的位置),距离短的位置更容易被取代,Ca2+比Mg2+更稳定。(为什么?,没有说明原因)

4.3. Substitution mechanism III: Np5+ + P5+ ? U6+ + Si4+
这应该是层内取代,
每个晶胞中两个四面体位置可以被取代,铀酰链上,在[010]方向聚合,连接由边和角共享硅四面体。耦合的P5取代是Si4位置为边和角与镎酰共享,NP - P的距离为3.17。

计算过程中,不能仅仅考虑取代阳离子的位置,NP的位置也有两种,对阳离子取代位置也有影响。
5 讨论
5.1参考相的比较
直接比较boltwoodite和镎标记过的boltwoodite总能不能提供融入的热力学信息,因为其化学组成、结构不同。可以比较两相分别的形成能。
K2O + 2UO3 + 2SiO2 + 4H2O ? K2(UO2)2(SiO3OH)2(H2O)3
ΔEformation = 466.97 eV
K2(UO2)2(SiO3OH)2(H2O)3 + NpO2 + ?O2 + ?H2O ?
HK2(UO2)(NpO2)(SiO3OH)2(H2O)3 + UO3
ΔEincorporation = 0.30 eV
反应物和产物的选择(source and sink)、环境条件、取代元素、计算近似方法(团簇或者周期性)等对融合能计算结果影响巨大。
采用NPO2作为源,UO3作为sink时,反应的融合能最低,因为采用2 NpO2 + ? O2比Np2O5更不稳定。因此,给融合反应提供了一个更大的推动力。实际考虑采用NP2O5,因为这种化合物更稳定。
二元氧化物从逻辑上来说对NP源及Usink的选择较好,但对于耦合取代来说,它们不是最好的选择,层间取代的机制来说,源采用CaO,sink采用K2O,这些氧化物的单胞较小,有利于量子力学计算,但这些氧化物在自然界中不普遍存在,相对于其它矿物来说更加不稳定,在环境中不太可能存在的阳离子源。因此,层间取代机制研究采用的是自然界分布比较广泛的硅酸盐矿物。(源)CaAl2Si2O8,sink为KAlSi3O8,
K2(UO2)2(SiO3OH)2(H2O)3 + ?Np2O5 + CaO ?KCa(UO2)(NpO2)(SiO3OH)2(H2O)3 + UO3 + ?K2O
ΔEoxides = 2.40 eV
K2(UO2)2(SiO3OH)2(H2O)3 + ?Np2O5 + CaAl2Si2O8 + SiO2 ?
KCa(UO2)(NpO2)(SiO3OH)2(H2O)3 + UO3 + KAlSi3O8 + ?Al2O3
ΔEsilicates = 1.20 eV
层内取代机制:
K2(UO2)2(SiO3OH)2(H2O)3 + ?Np2O5 + ?P2O5 ?
K2(UO2)(NpO2)(SiO3OH)(PO3OH)(H2O)3 + UO3 + SiO2
ΔEoxides = -1.10 eV
融合能是负值,表明,如果发生的是层内取代,NP可以无限的融合到boltwoodite中。但固溶计算表明,boltwoodite融入的NP是有限的。如果把源改成berlinite (AlPO4),sink为corundum (Al2O3)的话,融合能为0.86eV。这与环境条件比较相符合。因此,特定环境中的相考虑是十分必要的。实际的环境中更可能是水复合物,例如,在地下水容易形成含有低浓度锕系元素的碳酸盐,计算中也做了考虑,H渗入机制中选了简单的水溶液,如(NpO2+aq)。 周期性量子力学计算中,带电体系的计算很难处理,可以采用团簇类型的量子力学计算方法,但是同一个化学反应中能量的计算应该采用一致的方法,否则无法比较,解决这个问题的一个方法是组成由分反应构成的整体的反应方程式,反应使用一致的计算方法,但不同子反应可能是不同的计算办法,分别是处理最适合的特定的边界条件。一些固体或有限分子可携带的电荷。中性采用CASTEP计算,带电使用Dmol3计算。
(这种方法计算出来的数据是否可信?有没有其它的例子来说明这个问题?)
5.2. Limit of Np-incorporation
这个是怎么计算的不是太明白?
5.3.Impact of boltwoodite chemistry on Np incorporation
离子半径用来评估固溶物的热力学稳定性,离子半径相近的离子间容易取代。
5.4 反应机制的可行性
从反应机制的可行性来说,不仅仅要考虑热力学能量,还应该考虑空间几何结构等因素,H+可能是最有可能发生交换反应的,动力学瓶颈在于H3O+的形成。
个人觉得作者应该不是美国人,英文写得还是有点难看懂,在语法上总感觉很多地方别扭,
是否做些修改呢?