原核表达步骤
原核表达先要将基因克隆到原核表达载体上,然后通过转化到JM109或BL21等菌株中,诱导表达蛋白,然后进行蛋白纯化。本实验方案的前提是,目的基因已克隆到载体,并已转进入JM109菌株中。
一.鉴定目的蛋白是否在大肠杆菌JM109或BL21中大量表达
(一)制样
1 . 挑取经过双酶切鉴定的单克隆菌落于700ul LB培养基,加入0.7ul Amp(100mg/mL),37oC200r/min摇床培养,过夜活化。
2. 以1:50比例(200ul),将活化的过夜培养物加入10mL LB液体培养基中,加入10uLAmp(100mg/ml),37oC200r/min摇床扩大培养2h-3h,期间取样监控菌液的OD值,控制菌液OD600在0.6-1.0之间,以使大肠杆菌处于最适合表达外源蛋白的生长状态。(一般3h时,菌液浓度及达到标准,但是不同的基因对菌的影响不同,所以第一次实验时需要确定这个最佳时间)
3. 从10ml扩大培养物中取3ml菌液作为不加IPTG的空白对照(CK),其余7ml菌液加入7ul IPTG(储存浓度为0.5mol/l),使IPTG终浓度达到0.5mmol/l。以200r/min的转速,37oC摇床培养3h。
4. 以5000r/min离心2min收集菌体,倾倒上清,每个离心管收集3ml培养物。
5. 加入1ml dH2O,将管底沉淀用振荡器打散以充分洗涤,8000r/min离心2min,倾倒上清。
6. 重复步骤5。将离心管中的水倒干净。
(二)菌落SDS-PAGE
1. 在收集的菌体中加入200ul 1×SDS PAGE loading buffer(可根据沉淀的量增加或减少loading buffer的量,一般200ul比较合适)。用漩涡器剧烈震荡,确保将管底沉淀震散。
2. 将样品于100℃恒温加热器上开盖加热10min(Marker也要加热)。样品凉后,12000r/min离心3min,取每管的上清点样。上样量一般30ul—40ul,marker 20ul。
(三)SDS-PAGE分析
1. 根据目的片段的大小,制作不同浓度的分离胶
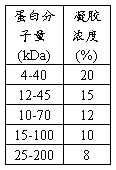
分离胶(两块)配方
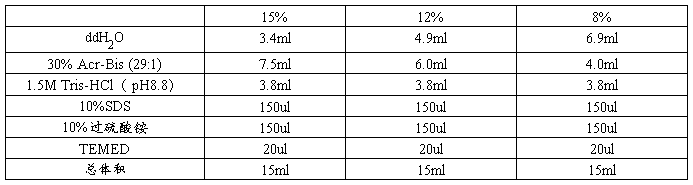
按表中所列顺序从上到下加试剂,加完TEMED后,轻轻摇匀液体,一块胶加7ml溶液。加完后, 用1mLddH2O封住分离胶液面,在室温(37℃)凝结30min,至分离胶与水之间出现一条清亮的分界线,倾倒水层,用滤纸条将残余的水吸干。
2.制作浓缩胶
浓缩胶(两块)配方
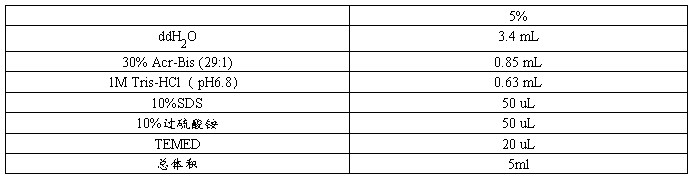
胶配好后混匀,灌胶2ml,插梳子时根据点样量和浓缩胶长短调整梳子插入的深度,于室温(37℃)凝胶30min。
3.胶凝好后,拔梳子,将胶放入电泳槽中,加入1×Tris-Gly电泳缓冲液没过点样孔,胶外侧缓冲液界面应完全没过电极
4.Marker点5uL,用10uL小枪点样,以防样品冲出点样孔与旁边样品混杂。
电压打到70V,保证电流在20mA-30mA之间,当样品跑入浓缩胶时可适当调高电压,但不宜多次调整电压,否则条带会跑歪。
5.当溴酚蓝电泳至胶槽底部时,一般需要2.5h,停止电泳,剥胶,将浓缩胶部分切除后,加入考马斯亮蓝染色液,40r/min染色3h,可以过夜染色。
6.用移液器回收考马斯亮蓝染色液,再用蒸馏水将浮色冲洗干净后,倒入考马斯亮蓝脱色液,40r/min脱色至胶背景透明,期间可每2h更换一次脱色夜。
根据菌落SDS-PAGE的结果,可以看到目的蛋白是否大量表达。
二.鉴定目的蛋白形成的是包涵体还是可溶性蛋白
1. 从冻存的菌液中取10ul,接种于5ml的LB培养基中,加入5ul Amp(100 mg/ml),过夜活化菌株。
2. 按照1:50的比例,从活化的菌株中,取1ml菌液接种于50ml LB培养基中,加入50ul Amp(100 mg/ml),扩大培养3 h。
3. 扩大培养后的菌液,取出10ml,不加IPTG,作为空白对照。剩余的40ml菌液中加入40ul IPTG(0.5mol/l),诱导蛋白表达。两者在37℃,200rpm的条件下培养3h。
4. 用50ml 离心管收集菌体,8000rpm,离心3min。注意配平。
5. 倒掉上清,加入40ml左右的dH2O洗菌体一遍,12000 rpm,离心2min。注意要将菌体充分打散。
6. 倒掉上清,用枪头将上清吸干净。根据菌体的浓度,加入适量的PBS buffer 悬浮菌体。
7. 用大功率的超声波将菌体破碎,其间超声波每工作2min,停止1min。菌体要始终保持在冰水混合物上,以保证低温,蛋白不易变性。如此反复6-10遍,直至菌液清澈。
8. 12000rpm, 离心3min,分离上清和沉淀。
9. 在沉淀中加入250ul 1×SDS Loading buffer ,取200ul 上清,加入50 ul 5×SDS Loading buffer,在恒温加热器上100℃加热10min。
10.加热后的样品冷却到室温后,12000rpm,离心2min。
11.取上清40ul 点样,按照一中的方法进行SDS-PAGE检测。点样顺序是:对照的上清,沉淀,样品的上清,沉淀。
12.根据SDS-PAGE结果,判断目的蛋白是以包涵体还是以可溶性蛋白的形式产生。如果蛋白在沉淀中大量表达,则形成包涵体,需要改变诱导表达条件,比如将扩大培养与诱导培养的温度改为25℃,并延长相应培养时间。如果蛋白在上清中表达,则形成可溶性蛋白,可继续进行下一步实验。
三.纯化目的蛋白(整个过程要尽量保持在4℃进行)
1. 大量诱导目的蛋白(一般需要2-4 L的菌量),用超声波破碎细胞,收集上清。
2. GST纯化柱的柱床保存在20%的乙醇中,使用之前,先用吸管将柱床轻轻加入到柱子中,打开柱子,流出乙醇,使柱床沉积下来。
3. 加入10ml PBS buffer,洗柱床3遍,使得柱床重新平衡。
4. 将准备好的样品加入到柱子中,每次加入5-10ml样品,使样品结合在柱子上(每次上样量不能太多,因为柱子的结合能力有限)。
5. 用PBS buffer洗柱子3遍,每次用10ml,去除柱子中非特异性结合的杂蛋白。
6. 加入5ml洗脱液(10m mol/l的还原型谷胱甘肽,用50mmol/l tris-bufferr或者PBS buffer溶解)将目的蛋白洗脱出来。用1.5ml离心管收集流出来的样品,每管接0.5ml样,一共收集6-8管(一般情况,第一管流出的是PBS,第二和第三管大部分是目的蛋白,所以OD值很大,第三至第六管的OD值慢慢降低。不要偷懒一次接太多,不然会影响后续收集蛋白。还原型谷胱甘肽很容易被氧化,如果一次没有用完,请用保鲜膜封口置于4度冰箱保存,超过2天的话请更换新的洗脱液。)
7. 用PBS buffer洗柱子1遍。
8. 用分光光度计测每一管的OD值,记录下读数,将读数大于200的样品留下(一般是第2至第4管),冻于-20℃冰箱中。
9. 重复步骤4-8,直至所有样品均纯化完毕。
10.可以用SDS-PAGE检测纯化出来的样品浓度以及纯度。
重生柱子的方法:(柱子使用三四次之后,需要重生,即让柱子上的GST结合基团重新暴露出来)
1.用还原型谷胱甘肽洗脱液洗柱子3遍,每次加入10ml。
2. 用 PBS缓冲液洗柱子4遍,每次加入10ml。
3. 用50 mmol Tris HCl(pH 8.0)洗柱子3遍,每次加入10ml。
4.用0.5M NaCl(pH 8.0)洗柱子3遍,每次加入10ml。
5. 用100 mM醋酸钠(pH 4.5)洗柱子3遍,每次加入10ml。
6. 用0.5M NaCL洗柱子3遍,每次加入10ml。
柱子长时间不用,需要按照以下方式清洗及保存:
1. 按照重生的1-6步骤先将柱子重生一遍。
2. PBS洗柱子2遍,每次加入10ml。
3. 将柱床用吸管吸出,分别用5%,10%,15%,20%的乙醇洗2遍。
4. 用单蒸水轻轻冲洗柱子底部的膜,洗出杂蛋白。
5. 柱床保存在含20%乙醇的PA瓶中。柱子和柱床置于4度冰箱保存。
如果柱子用于纯化不同的蛋白, 只需要按照重生方法的1,2步骤操作即可,然后直接纯化另外一个蛋白。如果柱子流速很慢,可能是柱子底部的膜被杂蛋白堵住了。柱子重生之后,将柱床用吸管吸出,然后用单蒸水将柱子底部的膜轻轻冲洗,再将柱床加入到柱子中,重新上样。
如果柱子需要重生,请用完柱子的同学自觉完成这份工作,以免影响下一位同学的使用。
四.根据不同的用途浓缩蛋白
1. 若蛋白用于制作抗体
如果洗脱液用的是tris-bufferr配制的,需要用1/4的PBS buffer进行透析,然后在冷冻干燥机中冻干浓缩,因为公司最终需要将目的蛋白溶于PBS buffer中。
如果洗脱液是用PBS buffer配制的,则直接在冷冻干燥机中冻干浓缩。
GST 纯化柱手册上建议的是用tris-bufferr配制洗脱液,但用PBS buffer配制的洗脱液可以将目的蛋白洗脱下来。可自己选用。
2. 若蛋白用于活性检测
由于蛋白在tris-bufferr和PBS buffer中,均能保持活性,所以不论用哪种,都不需要透析,直接浓缩干燥即可。
第二篇:原核表达总结
原核表达之目的基因克隆
1. 了解实验课题对目的蛋白的要求,包括:目的蛋白分子量有多大,表达目的(是蛋白结
晶、药剂结合还是制备抗体,不同目的对蛋白要求不同);是否要其可溶;是胞内表达还是分泌表达,是组成型表达还是诱导型表达;另外,还要了解蛋白表达后需要采用什么样的方式进行纯化,纯化标签有多大,蛋白纯化后是否需要将标签去除(即标签的存在是否会影响蛋白的活性)。
还要对目的蛋白进行稀有密码子(仅限于真核生物基因通过原核表达系统表达时参考,注意稀有密码子的多少,位置5位或3位,稀有密码子是否成串出现等)和可溶性网上预测等。
稀有密码子预测网址: http://molbiol.edu.ru/eng/scripts/01_11.html
原核表达蛋白可溶性预测网址: http://www.biotech.ou.edu/
综合以上,选取合适的表达载体及宿主菌(做原核表达前对各种载体及宿主菌的充分了解是必须的,建议首先应阅读《默克原核表达宝典》)。
注意:不是所有的标签都是用来纯化蛋白的,有的标签有促进蛋白溶解的作用,有的则是二硫键形成或利于表达后蛋白的检测。
一般我们最常用的是胞内诱导型表达(即蛋白翻译后是存在于细胞内,通过加诱导物的方法来控制表达水平)。
2. 搜索要表达的目的基因的序列,根据其编码区序列来设计引物;
注意:要注意在引物上加入限制性内切酶识别位点(首先应分析蛋白编码区内是否含有相同的内切酶识别位点),并通过查阅资料看两种酶(上下游引物分别加入酶切位点)是否可以在同一反应体系中起作用,并注意标签序列是在N端还是在C端,若要在C端保留标签,则需要将下游引物的终止密码子替换掉;若要在引物上引入标签序列,则引物上应加入标签序列碱基(即在上游引物或下游引物处引入His的密码子);另外,还要注意引物不能造成蛋白的编码框发生改变;
1,2步的分析至关重要,只有在完成前两步的工作后才可以进行后续的实验操作。
3. 摇菌,进行RNA抽提(注意选取不同表现型的菌株),摇菌的时间一定不要太长,太长
的话RNA活性不高;
4. mRNA反转成cDNA(此步应在第三步完成后立即操作,RNA存放时间长易降解);
5. 以cDNA为模板,利于设计好的引物进行PCR扩增;
6. 通过胶回收试剂盒回收目的DNA片段。
蛋白质表达载体构建步骤08.4.20(08.11.10日修改)
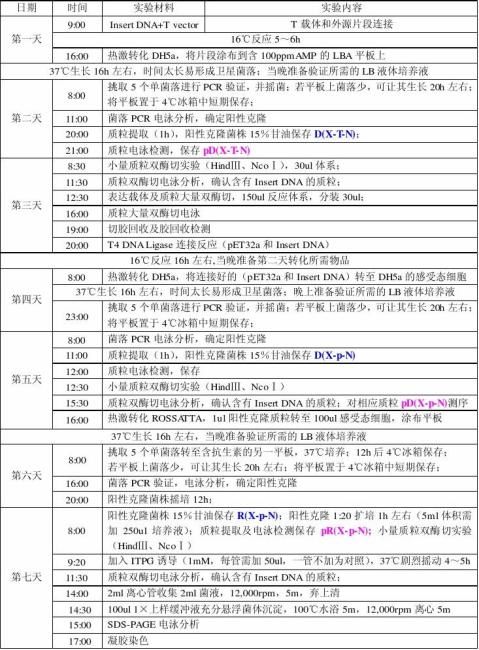

微管蛋白的可溶性表达及纯化(08.11.4日修改)
1. 将重组质粒(BL21-2β2或Rossatta-2β2)的表达菌37℃摇培过夜后,1:20
扩配(约需1h15m);
2. 至OD600=0.5~0.7时(约1h15min~1h45min),在15-(0.5-24,0.8-12);20-
(0.5-12,0.8-8);28、25-(0.8-8)进行蛋白诱导表达,具体条件根据摇床使用情况而定;
3. 对诱导后的菌液4℃,4000g,20min,彻底去上清;离心前取2ml以备用于
总蛋白电泳分析S1;
4. 细菌沉淀用BindingBuffer (无尿素,20mM咪唑)悬浮,100ml用5~10ml
缓冲液(约当2~5ml每克菌量),视悬浮后的菌液浓度而定;
5. 反复冻融法破碎细胞:做到14步时由于时间原因不能立即继续后面的操作,
可将菌悬液放于-70℃冰箱中冷冻;也可用反复冻融的方法来破碎细胞,在-70℃冻融,在室温下溶解,反复冻融3~4次;
6. 加Lysozyme至1mg/ml (1ml加10ul),在冰上孵育30min~1h;
7. 200~300W超声破碎,2s×30次,停顿2s;若菌种较多可以增加破碎次数。
注意超声破碎时间,不可太长,否则温度升高易导致蛋白变性;而时间太短则破碎不彻底,蛋白无法释放出来;
注意:第7步可选作,若经过冻融及溶菌酶处理后的菌悬液比较澄清,则可不用超声破碎。
8. 4℃,10000rpm 20~30min,取上清;将沉淀用变性的BindingBuffer (8M
尿素)悬浮,然后室温摇动30min,取40ul进行电泳样制备S2;
9. 4℃,10000rpm 10min,对上清再离心一次,取40ul进行电泳样制备S3;
10. 上清通过0.45um的细菌过滤器,取40ul进行电泳样制备S4;
11. 用HisTrap纯化:
a) 洗柱:用5倍柱床体积(5ml)纯水洗柱,避免产生气泡。
b) 平衡:至少5ml BindingBuffer (20mM咪唑),一般10ml,流速在
1ml/min。
c) 上样:流速要慢要,控制在1ml/min,并收集流出的蛋白液,取40ul进
行电泳样制备S5;
d) 重新上样:对第一次上样流出的蛋白液再进行过柱一次,取40ul进行电
泳样制备S6;
e) 平衡:用10ml BindingBuffer (20mM咪唑),收集第一管及最后一管,
各取40ul进行电泳样制备S7,S8。
f) 洗脱:用5ml ElutionBuffer (500mM咪唑)洗脱,每1ml一管,一般第
一二管中蛋白多。
g) 平衡:用5ml的BindingBuffer (20mM咪唑)平衡柱子;
h) 封闭:用20%的乙醇封闭柱子,4℃保存。
若需进行咪唑浓度优化,则纯化步骤改为:
10.4 梯度咪唑浓度洗脱:依次用20、30、40、50、75、100、150、200、300、400、500mM的咪唑进行洗脱,每个浓度各用4ml,收集第2、3V;电泳分析后确定最佳的咪唑平衡及洗脱浓度。
12. 样品处理:吸取纯化后的蛋白液40ul,加入10ul的5×的上样缓冲液,100℃水浴5min,12,000rpm离心3min后取上清点样,每个加样孔15ul。
13. 80V电泳2h左右,取出胶用染色液染色4~5h(回收的染色液可过夜),用脱色液脱色3次,观察拍照。
附:
1. 总蛋白电泳方法:700ul,12000g离心5min,弃上清,加入50μL 1×蛋白上样缓冲液,充分悬浮,煮沸5min,12000g离心5min;
2. 构建好的载体在进行蛋白诱导表达前,应选取10个左右的单克隆进行蛋白诱导的小量试验,对诱导后的总蛋白进行电泳分析,选取表达量最大的克隆保存(甘油菌,-70℃冰箱);
3. 从-70℃冰箱中活化菌进行蛋白诱导表达试验时,活化好的表达菌在4℃冰箱仅可存放15天左右,最多1个月,过期后必须重新划线活化。4℃冰箱保存的宿主菌每次用过后必须用封口膜封好。
4. SDS-PAGE分析的蛋白样:总蛋白样S1,沉淀中的蛋白样S2,离心后上清中的蛋白样S3,过细菌过滤器后的蛋白样S4,第一次上柱时流出的蛋白样S5,第二次上柱时流出的蛋白样S6,20mM Bingding buffer过柱后的第一管S7和最后一管蛋白样S8。