实验二 熔点的测定及温度计校正
一. 实验目的:
1. 了解熔点测定的原理及意义;
2. 掌握熔点测定的基本操作方法;
二. 实验重点和难点:
1. 熔点测定的意义;
2. 熔点测定的操作方法;
实验类型:基础性 实验学时:4学时
三. 实验装置和药品:
主要实验仪器:熔点管;表面皿;玻璃棒;长40cm的玻管;
Thiele管(又称b形管);酒精灯;温度计;液体石蜡;
主要化学试剂:苯甲酸(熔点mp122.40C);未知样品(或者尿素):水杨酸(mp1590C)
或乙酰苯胺(mp114.30C)
四. 实验装置图:

五. 实验原理:
1.熔点 熔点是固体有机化合物固液两态在大气压力下达成平衡的温度,纯净的固体有机化合物一般都有固定的熔点,固液两态之间的变化是非常敏锐的,自初熔至全熔(称为熔程)温度不超过0.5-1℃。物质受热后,从开始熔化到全部熔完的温度差称作熔点距(或熔程),纯化合物的熔点距△≤0.5~1℃,据此,可根据熔点测定初步鉴定化合物或判断其纯度。
加热纯有机化合物,当温度接近其熔点范围时,升温速度随时间变化约为恒定值,此时用加热时间对温度作图(如图1)。
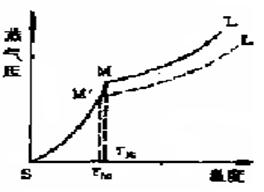

图1 相随时间和温度的变化 图2 物质蒸气压随温度变化曲线
化合物温度不到熔点时以固相存在,加热使温度上升,达到熔点.开始有少量液体出现,而后固液相平衡.继续加热,温度不再变化,此时加热所提供的热量使固相不断转变为液相,两相间仍为平衡,最后的固体熔化后,继续加热则温度线性上升。因此在接近熔点时,加热速度一定要慢,每分钟温度升高不能超过2℃,只有这样,才能使整个熔化过程尽可能接近于两相平衡条件,测得的熔点也越精确。
当含杂质时(假定两者不形成固溶体),根据拉乌耳定律可知,在一定的压力和温度条件下,在溶剂中增加溶质,导致溶剂蒸气分压降低(图2中M´L´),固液两相交点M´即代表含有杂质化合物达到熔点时的固液相平衡共存点,TM´为含杂质时的熔点,显然,此时的熔点较纯粹者低。
2.混合熔点
在鉴定某未知物时,如测得其熔点和某已知物的熔点相同或相近时,不能认为它们为同一物质。还需把它们混合,测该混合物的熔点,若熔点仍不变,才能认为它们为同一物质。若混合物熔点降低,熔程增大,则说明它们属于不同的物质。故此种混合熔点试验,是检验两种熔点相同或相近的有机物是否为同一物质的最简便方法。多数有机物的熔点都在400℃以下,较易测定。但也有一些有机物在其熔化以前就发生分解,只能测得分解点。
六. 实验內容及步骤:
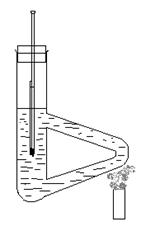
1.安装测定装置和取样 :【参阅教材P42图2.4】
(1) .熔点测定装置包括温度计、毛细管、Thiele管。
(2) .将毛细管一端在酒精灯上转动加热,烧融封闭。取干燥、研细的待测物样品放在表面皿上,将毛细管开口一端插入样品中,即有少量样品挤入熔点管中。然后取一支长玻璃管,垂直于桌面上,由玻璃管上口将毛细管开口向上放入玻璃管中,使其自由落下,将管中样品敦实。重复操作使所装样品约有2-3mm高时为止。
(3) .向Thiele管中加入液体石蜡(作为加热介质)直到支管之上。在温度计上附着一支装好样品的毛细管,毛细管中样品与温度计水银球处于同一水平。将温度计带毛细管放入Thiele管中,使温度计水银球位置在Thiele管中部。
将少许样品放于干净表面皿上,用玻璃棒将其研细并集成一堆。把毛细管开口一端垂直插人堆集的样品中,使一些样品进入管内,然后,把该毛细管垂宜桌面轻轻上下振动,使样品进人管底,再用力在桌面上下振动,尽量使样品装得紧密。或将装有样品,管口向上的毛细管,放入长约50一60cm垂直桌面的玻璃管中,管下可垫一表面皿,使之从高处落于表面皿上,如此反复几次后,可把样品装实,样品高度2—3mm。熔点管外的样品粉末要擦干净以免污染热浴液体。装入的样品一定要研细、夯实。否则影响测定结果。
2.熔点的测定:
(1) .在Thiele管弯曲部位加热。接近熔点(距熔点十几度)时,减慢加热速度,每分钟升1oC左右,接近熔点温度时,每分钟约0.2oC观察、记录晶体中形成第一滴液体时的温度(初熔温度开始塌陷并有液相产生)和晶体完全变成澄清液体时的温度(终熔温度)。
(2) .熔点测定应有至少两次平行测定的数据,每一次都必须用新的毛细管另装样品测定,而且必须等待液体石蜡冷却到低于此样品熔点20-30oC时,才能进行下一次测定。
(3) .对于未知样品,可用较快的加热速度粗测一次,在很短的时间里测出大概的熔点。实际测定时,加热到这个熔点以下10-15oC,必须缓慢加热,使温度慢慢上升,这样才可测得准确熔点。
按图搭好装置,放入加热液(浓硫酸或者液体石蜡),用温度计水银球蘸取少量加热液,小心地将熔点管粘附于水银球壁上,或剪取一小段橡皮圈套在温度计和熔点管的上部(如下图)。将粘附有熔点管的温度计小心地插入加热浴中,以小火在图示部位加热。开始时升温速度可以快些,当传热液温度距离该化合物熔点约10一15℃时,调整火焰使每分钟上升约1—2℃,愈接近熔点,升温速度应愈缓慢,每分钟约0.2一0.3℃。为了保证有充分时间让热量由管外传至毛细管内使固体熔化,升温速度是准确测定熔点的关键;另一方面,观察者不可能同时观察温度计所示读数和试祥的变化情况,只有缓慢加热才可使此项误差减小。记下试样开始塌落并有液相产生时(初熔)和固体完全消失时(全熔)的温度读数,即为该化合物的熔距。要注意在加热过程中试祥是否有萎缩、变色、发泡、升华、碳化等现象,均应如实记录。
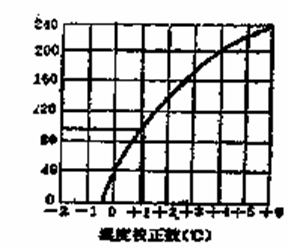
3.温度计校正
测熔点时,温度计上的熔点读数与真实熔点之间常有一定的偏差。这可能由于以下原因,首先,温度计的制作质量差,如毛细孔径不均匀,刻度不准确。其次,温度计有全浸式和半浸式两种,全浸式温度计的刻度是在温度计汞线全部均匀受热的情况下刻出来的,而测熔点时仅有部分汞线受热,因而露出的汞线温度较全部受热者低。为了校正温度计,可选用纯有机化合物的熔点作为标准或选用一标准温度计校正。
选择数种已知熔点的纯化合物为标推,测定它们的熔点,以观察到的熔点作纵坐标,测得熔点与已知熔点差值作横坐标,画成曲线,即可从曲线上读出任一温度的校正值。
七. 实验注意事项:
(1) 要测得准确熔点,样品一定要研得极细,装得结实,使热量的传导迅速均匀.
(2) 掌握升温速度是准确测定熔点的关键。这一方面是为了保证有充分的时间让热量由管外传至管內,以使固体熔化;另一方面因实验者不能同时观察温度计所示度数和样品的变化情况。只有缓慢加热,才能使此项误差减小。
(3) 注意橡胶塞子一定要开口,否则易产生暴沸现象。
(4) 温度计插入开口塞子中,刻度应面向塞子开口,其水银球位于b形管上下两叉管口之间。
(5) 熔点管必须洁净。如含有灰尘等,能产生4—10OC的误差。
(6) .熔点管底未封好会产生漏管。
(7) .样品粉碎要细,填装要实,否则产生空隙,不易传热,造成熔程变大。
(8) .样品不干燥或含有杂质,会使熔点偏低,熔程变大。
(9) .样品量太少不便观察,而且熔点偏低;太多会造成熔程变大,熔点偏高。
(10) .升温速度应慢,让热传导有充分的时间。升温速度过快,熔点偏高。
(11) .熔点管壁太厚,热传导时间长,会产生熔点偏高。
(12) .使用硫酸作加热浴液要特别小心,不能让有机物碰到浓硫酸,否则使浴液颜色变深,有碍熔点的观察。若出现这种情况,可加人少许硝酸钾晶体共热后使之脱色。采用浓硫酸作热浴,适用于测熔点在220℃以下的样品。若要测熔点在220℃以上的样品可用其它热浴液。
八. 实验相关內容:
熔点是有机物的重要物理常数之一。熔点是纯净有机物晶体熔融时的相变温度,纯净的有机物晶体有唯一确定的熔点。
测定有机物的熔点可以帮助推断未知的有机物。对已知的有机物可以根据所测的熔点与文献值推测其是否纯净。
了解有机物熔融过程的相平衡,了解有杂质存在时熔点的变化趋势,有助于理解熔点测定的现象,了解其应用。
九. 思考题:
1.测熔点时,若有下列情况将产生什么结果? (1)熔点管壁太厚。 (2)熔点管底部未完全封闭,尚有一针孔。 (3)熔点管不洁净。 (4)样品未完全干燥或含有杂质。 (5)样品研得不细或装得不紧密。 (6)加热太快。
第二篇:实验二 熔点的测定
实验二 熔点的测定
一、实验目的
1.了解熔点测定的意义。
2.掌握熔点测定的操作方法。
3.了解利用对纯粹有机化合物的熔点测定校正温度计的方法。
4.掌握热浴间接加热技术。
二、实验原理
晶体化合物的固液两态在大气压力下成平衡时的温度称为该化合物的熔点。纯粹的固体有机化合物一般都有固定的熔点,即在一定的压力下,固液两态之间的变化是非常敏锐的,自初熔至全熔(熔点范围称为熔程),温度不超过0.5-1℃。如果该物质含有杂质,则其熔点往往较纯粹者为低,且熔程较长。故测定熔点对于鉴定纯粹有机物和定性判断固体化合物的纯度具有很大的价值。

如果在一定的温度和压力下,将某物质的固液两相置于同一容器中,将可能发生三种情况:固相迅速转化为液相;液相迅速转化为固相;固相液相同时并存。
上图(1)表示该物质固体的蒸气压随温度升高而增大的曲线;
上图(2)表示该物质液体的蒸气压随温度升高而增大的曲线;
上图(3)表示(1)与(2)的加合,由于固相的蒸气压随温度变化的速率较相应的液相大,最后两曲线相交于M处(只能在此温度时),此时固液两相同时并存,它所对应的温度TM即为该物质的熔点。
上图(4)当含杂质时(假定两者不形成固溶体),根据拉乌耳(Raoult)定律可知,在一定的压力和温度条件下,在溶剂中增加溶质,导致溶剂蒸气分压降低(图中M1L’1),固液两相交点M1即代表含有杂质化合物达到熔点时的固液相平衡共存点,TM1为含杂质时的熔点,显然,此时的熔点较纯粹者低。
三、课堂内容
1.通过提问检查学生预习情况(CAI课件预习及实验书预习),提问内容如下:
(1)什么是熔点? 测定熔点有何意义?
(2)什么是熔程?纯净的固体有机化合物的熔程是多少?物质不纯时熔点及熔程有何变化?
(3)测熔点时样品为什么要研细,装实?
(4)油浴温度下降多少时再换另一根样品管?
(5)熔点管中装样品多少?
(6)当接近熔点时加热速度为多少?
(7)做本实验时应注意什么?
2.让学生演示安装及操作并讲解。
3.教师讲解熔点测定仪的使用与操作方法及注意事项。
四、课堂学生测试的样品

五、仪器、药品
仪器:①Thiele管;②200℃温度计;③熔点管、长玻璃管(60cm);④表面皿(中号)、锉刀、切口软木塞、胶塞、橡皮圈⑤数字熔点仪、镊子等。⑥酒精灯
药品:未知物1苯甲酸;未知物2水杨酸;浓硫酸(热载体);工业酒精
六、实验步骤
1.熔点管的制备(参见相关实验)
2.向熔点管填装样品
样品粉末要研细,取少许研细干燥的待测样品于表面皿上,并聚成一堆。将熔点管开口向下插入样品中,然后将熔点管开口向上轻轻在桌面上敲击,使样品进入熔点管,再取约40cm的干净玻璃管垂直台面上,把熔点管上端自由落下,重复几次使样品填装紧密均匀,以达到测熔点时传热迅速均匀的目的。样品高度约2-3mm。装好后去除沾在熔点管外的样品,以免沾污热浴。一次熔点测定一般同时装好4根熔点管。对于易升华或易吸潮的物质,装好后立即把毛细管口用小火熔封。
3.热浴的选择与装配仪器
根据样品的熔点选择合适的加热浴液。加热温度在140℃以下时,选用液体石蜡或甘油。温度低于220℃时可选用浓硫酸,因硫酸具有较强的腐蚀性,所以操作时应注意安全。
向b形管中加入浴液至支管之上1cm处。然后固定在铁架台上;毛细管底部应位于温度计水银球中部,用橡皮圈或线绳将毛细管固定在温度计上,橡皮圈不能浸入浴液;用b形管时,温度计的水银球应处于两侧的中部,不能接触管壁。
4.校正温度计
将已知样品放入热浴中,测定其熔点,将测定值与文献值对照,其差值即为校正系数。
5.测定
(1)粗测
若未知物的熔点,应先粗测一次。粗测加热可稍快,升温速率约5-6℃/min。到样品熔化,记录熔点的近似值。
(2)精确测定
测定前,先待热浴温度降至熔点约30℃以下,换一根样品管,慢慢加热,一开始5℃/min,当达到熔点下约15℃时,以1-2℃/min升温,接近熔点时,以0.2-0.3℃/min升温,当毛细管中样品开始塌落和有湿润现象,出现下滴液体时,表明样品已开始熔化,为始熔,记下温度,继续微热,至成透明液体,记下温度为全熔。
①熔点测定至少要进行2次,2次的数据应一致。每次测定必须用新装样品的毛细管,不可用已测过熔点的样品管。对于未知熔点的测定,可先以快速加热粗测,然后待浴液冷却至熔点以下约30℃左右时,再做精密的测定。
②通常在熔程为0.5-1.0℃时化合物则被认为是纯物质。
③物质熔点测定是有机化学工作者常用的一种技术,所得的数据可用来鉴定晶体的有机化合物并作为该化合物纯度的一种指标。
七、装置图
八、注意事项
1.实训完毕,待浴液冷却后,将浴液倒回原瓶中,用滤纸擦去温度计上的浴液冷却后,用水冲洗,以免温度计水银球破裂。
2.样品要研细、装实,使热量传导迅速均匀。样品高度2-3mm,沾附于管外粉末必须擦去,以免污染加热浴液。
3.控制升温速度,开始稍快,当传热液温度距离该化合物熔点约10℃时,调整火焰上升速度为1-2℃/min,愈接近熔点,升温速度愈慢,使0.2-0.3/min。
4.仪器的装配一般是从下向上,从左到右,先难后易逐个地装配,拆卸时,按照与装配时相反的顺序,逐个拆除。
5.仪器用铁夹牢固地夹住,不宜太松或太紧,铁夹不能与玻璃直接接触,应套上橡皮管,垫上石棉网,需要加热的仪器,应夹住仪器受热最低的位置,冷却管则应夹在中间部分。
6.每个样品测三次(粗测一次,细测两次)。
7.注意观察温度计的温度。
8.油浴温度下降30℃以下时更换另一根管。
9.用熔点测定仪测熔点时,取放盖玻片和隔热玻璃时,一定要用镊子夹持,严禁用手触摸,以免烫伤(因熔点热台属高温部件)。
10.温度超过250℃时,浓硫酸发生白烟,防碍温度的读数,在这种情况下,可在浓硫酸中加入硫酸钾,加热使成饱和溶液,然后进行测定。
11.在热浴中使用的浓硫酸有时由于有机物掉入酸内而变黑,防碍对样品熔融过程的观察,在这种情况下,可以加入一些硝酸钾晶体,以除去有机物质。
九、熔点测定可能产生误差的因素
1.熔点管不洁净。如含有灰尘等,能产生4-10℃的误差。
例如,熔点管内壁含有碱性物质,能催化醛或酮的羟醛缩合,或使糖类及其衍生物发生变旋作用,而使熔点降低,熔程变大;故熔点管必须洁净。
2.点管底未封好而产生漏管。检查是否为漏管,不能用吹气的方法,否则会使熔点管内产生水蒸气及其它杂质。检查的方法为:当装好样品的毛细管浸入浴液后,发现样品变黄或管底渗入液体,说明为漏管,应弃去,另换一根。
3.样品粉碎不够细。填装不结实,产生空隙,则不易传热,造成熔程变大。
4.样品不干燥或含有杂质。根据拉乌耳(Raoult)定律,会使熔点偏低,熔程变大。
5.样品量的多少也会影响。太少不便观察,产生熔点偏低;太多会造成熔程变大,熔点偏高。
6.升温速度应慢,让热传导有充分的时间。升温速度过快,熔点偏高。
7.熔点管壁太厚,热传导时间长,会产生熔点偏高。
归纳上述因素为四个:
(1)熔点管规范(包括规格、管底封闭、洁净等)。
(2)样品合格(包括干燥、粒度等)。
(3)样品填装符合要求(量的多少、填充结实与否)。
(4)升温速度。
十、实验要求
1.准确测试样品的熔点。
2.实验后上交实验报告。
十一、讨论题
1.测定熔点时产生误差的因素有哪些?
2.毛细管法与数字熔点仪法测熔点的优缺点各是什么?
3.两个样品的熔点相同,能否确定它们是同一种物质?
4.测定熔点时,下列情况对实验结果有何影响?
(1)加热过快 (2)样品中有杂质 (3)熔点管太厚
(4)熔点管不干净 (5)温度计未校正
5.若样品研磨的不细,对装样品有什么影响?对测定有机物的熔点数据是否可靠?
6.加热的快慢为什么会影响熔点?在什么情况下加热可以快一些,而在什么情况下加热则要慢一些?
7.是否可以使用第一次测定熔点时已经熔化了的有机化合物再做第二次测定呢?为什么?