实验一 槐米中芦丁的提取、精制及含量测定
一、实验目的
1.掌握水提取法提取黄酮苷的原理及方法。
2.掌握芦丁的精制方法
3.掌握分光光度法测定芦丁含量的方法
二、实验原理
芦丁可溶解于热水,利用不同温度条件下,物质的溶解度不同的特性,通过降低温度使芦丁的浓度达到饱和状态而结晶析出。
三、实验用品
仪器:粉碎机、样品筛、电炉、石棉网、布氏漏斗、滤纸、脱脂棉、真空泵、抽滤瓶、天平、圆底烧瓶、冷凝回流管、层析缸、毛细点样管、电吹风、显色喷瓶、分光光度计。
材料:槐花米。
试剂:蒸馏水、硫酸、乙醇、色谱纯甲醇、甲酸、芦丁对照品等。
四、实验方法
1.芦丁的提取
取槐米20 g,加沸水250 ml,煮沸提取30min,纱布或脱脂棉趁热过滤,残渣同法再操作一次,合并两次滤液。将滤液适当浓缩(原体积一半)后室温放置析晶,待全部析出后,减压抽滤,用冷蒸馏水洗涤芦丁结晶,抽干,得粗制芦丁(若抽滤堵滤纸可采用离心的方法:离心12000rpm,10min),置空气中干燥后,称重。芦丁得率(%)=粗制芦丁(g)÷粗粉重量(g)×100%。
2.芦丁的精制纯化
取粗制芦丁,按照1:60加入蒸馏水,煮沸15min,趁热抽滤,滤液冷却静置后即可析出晶体,抽滤,得芦丁精制品,置空气中干燥后,称重。
3.芦丁的水解(槲皮素的制备)
取精制芦丁适量,研细后置于250 ml圆底烧瓶中,按照1:70的量加入2%硫酸,加热回流40~60min,生成鲜黄色沉淀,放冷沉淀,抽滤,沉淀物为芦丁的苷元即槲皮素,用蒸馏水洗至中性,抽干水分,晾干,称重即为粗制槲皮素。粗制槲皮素再用95%乙醇回流溶解,趁热过滤,加蒸馏水至乙醇浓度为50%,重结晶得精制的含2分子结晶水的槲皮素。
4.芦丁和槲皮素的薄层层析
4.1点样:在距离薄层板下边缘2cm处用毛细管点样,原点直径不应超过2~3mm,原点之间距离约为1~2cm。本实验点1、2、3、4四个样点(1为精制的芦丁,2为芦丁标准品,3为粗制槲皮素,4为精制槲皮素)。
4.2展开:点样后选择合适的展开剂在层析缸内展开。展开剂的选择,可以参考文献中和自己研究的样品类似物质的分离,也可以在薄层板上每隔2~3cm距离点上样品溶液,待斑点上的溶剂挥发后,向各斑点上分别滴加不同的展开剂展开如图比较,A~D四种溶剂,溶剂B比较适当。单一的溶剂分离效果不佳时,可以调制混合溶剂进行实验。溶剂前沿接近薄层板上端时,取出薄层板停止展开。本实验中展开剂为乙酸乙酯-甲醇-水(8:1:1)展开,展距12cm。
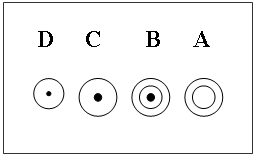
4.3显色:取出的薄层板记录溶剂前沿位置,挥发干展开剂后,用显色剂进行显色。在薄层板上喷雾10%~20%硫酸后,于100℃以上加热,就可以出现灰褐色斑点,或者在碘蒸气中薰,则出现褐色斑点。这两种显色方法几乎适用于全部有机化合物。本实验用碘蒸气显色后观察样品斑点的位置,并计算Rf值。
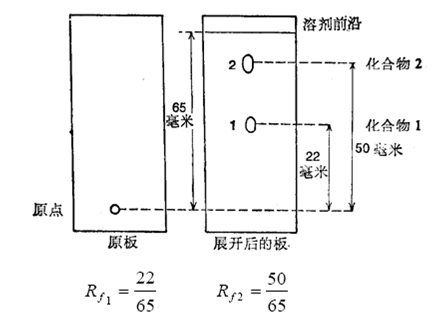
4.4计算Rf:原点至溶剂前沿距离为a,样品斑点中心至原点距离为b,则Rf=b/a 。一般鉴定纯化合物时,Rf值最好在0.4~0.6之间。被分离物质在展开剂中的Rf值之差最好大于0.05 。
5.槐米中芦丁含量测定
5.1色谱条件
色谱柱:十八烷基硅烷键合硅胶为填充剂的C18柱(25cm×4.6mm×5μm),以甲醇-1%冰醋酸水溶液(32:68)为流动相;流速为1.00ml/min;进样量为20μl;柱温为25℃;检测波长257nm。
5.2 标准曲线的制作
精确称取120℃真空干燥恒重的芦丁对照品7.9mg(或适量),用甲醇定容于50ml容量瓶中,摇匀,得对照品溶液。分别精密吸取对照品溶液0.50、1.00、2.00ml于10ml容量瓶中用甲醇定容,然后在上述色谱条件下进样,测定峰面积,以芦丁对照品浓度为横坐标,对应峰面积为纵坐标,进行线性回归绘制标准曲线。
5.3 槐米中芦丁含量的测定
精密称取槐米干燥粉末0.1g置于具塞锥形瓶中,精密加入甲醇50ml,称重,超声提取30min(功率250W,频率25kHz),冷却后称重,甲醇补足失重,摇匀过滤。精密量取续滤液2ml,置于10ml容量瓶中,加甲醇至刻度,摇匀。取适量样品溶液用0.45μm微孔滤膜过滤后作为供试样品液,进样20μl供试样品液进行色谱分析,计算峰面积,代入回归方程计算样品中芦丁的含量。
5.4 精制芦丁纯度的测定
精密称取精制芦丁0.1g置于具塞锥形瓶中,精密加入甲醇50ml,称重,超声提取30min(功率250W,频率25kHz),冷却后称重,甲醇补足失重,摇匀过滤。精密量取续滤液2ml,置于10ml容量瓶中,加甲醇至刻度,摇匀。取适量样品溶液用0.45μm微孔滤膜过滤后作为供试样品液,进样20μl供试样品液进行色谱分析,计算峰面积,代入回归方程计算样品中芦丁的含量,并计算精制芦丁样品中芦丁的纯度。
六、作业
1.计算热水提取芦丁的得率?
2.计算从粗制芦丁到纯化后芦丁的得率。
3.计算由芦丁水解产生粗制槲皮素的得率,乙醇结晶纯化后精制槲皮素的得率。
4.记录芦丁和槲皮素薄层色谱行为并计算Rf值。
5. 线性回归,绘制芦丁标准曲线通过标准曲线计算槐米中芦丁的含量和精制芦丁样品中的芦丁的纯度。
实验二 HPLC法测定甘草中甘草苷的含量
一、实验目的
1. 了解HPLC原理
2. 掌握HPLC测定甘草苷含量的方法
二、实验用品
仪器:岛津LC-20A高效液相色谱仪、岛津PDA检测器、 色谱工作站、赛多利斯万分之一天平。
材料:甘草饮片。
试剂:甲醇、冰醋酸、色谱纯乙腈、超纯水、甘草苷对照品等
三、实验方法
1.色谱条件
色谱柱:C18(25cm×4.6mm×5μm),流动相乙腈-0.5%冰醋酸(1 ∶ 4) ,流速0. 8 mL/min,检测波长276 nm,柱温室温,进样体积10 μL。
2.标准曲线的制作
精密量取甘草苷对照品2mg于10ml容量瓶,甲醇溶解配制200μg/ml对照品储备液,然后倍比稀释为50、20、10、5、1μg/ml(分别取200μg/ml对照品储备液2.5、1,0.5,0.25,0.05ml,20%乙腈定容至10ml),然后将200、50、20、10、5、1μg/ml对照品溶液分别进样10 μL。按色谱条件测定甘草苷峰面积。以对照品量为横坐标,峰面积为纵坐标,绘制标准曲线,得回归方程。
3.甘草供试样品的制备及其甘草苷含量的测定
精密称取甘草样品粉末0.4g于100ml锥形瓶中, 精密加入70%乙醇20ml,精密称定后,置于超声波清洗仪中提取30min,取出冷却至室温,称重,补足失去的溶剂量过滤后 精密量取滤液0.5ml于10ml容量瓶中,用20%乙腈稀释至刻度,摇匀,即得供试样品溶液C。
精密称取甘草样品粉末0.4g于150ml圆底烧瓶中, 精密加入70%乙醇20ml,精密称重后置于水浴回流提取30min,取出冷却至室温,称重,补足失去的溶剂量,抽滤。 精密量取滤液0.5ml于10ml容量瓶中,用20%乙腈稀释至刻度,摇匀,即得供试样品溶液H。
取适量不同方法制备的供试样品溶液(C和H),用0.45μm微孔滤膜过滤后取10μl进行色谱分析,计算峰面积,代入回归方程计算样品中甘草苷的含量。
四、作业
1.线性回归,绘制甘草苷标准曲线
2.通过标准曲线计算不同提取方法供试样品液中甘草苷的含量,并比较两种提取方法的优劣
实验三 超声、回流及压力快速溶剂三种提取方法提取葵花籽油
一、实验目的
掌握超声波提取、加热回流提取及压力快速溶剂提取法的原理及操作方法。
二. 实验原理
1. 超声波提取法
超声波振动产生并传递强大的能量,大能量的超声波作用在液体里,在振动处于稀疏状态时,液体会被撕裂成很多的小空穴,这些小空穴一瞬间即闭合,闭合时产生高达几千大气压(称为空化效应)。这种空化效应在溶剂内部产生强烈冲击波和速度极快的微射流,能使细胞壁破裂,使细胞里边的物质释放出来。在油脂的提取中加快了油脂的渗出速度,提高了出油率。
2.加热回流提取法
当溶剂加到原料中时,溶剂由于扩散、渗透作用逐渐通过细胞壁透入到细胞内,溶解了可溶性物质,而造成细胞内外的浓度差,于是细胞内的浓溶液不断向外扩散,溶剂又不断进入药材组织细胞中,如此多次往返,直至细胞内外溶液浓度达到动态平衡时,将此饱和溶液滤出。回流提取采用回流加热装置,在水浴中加热回流,以免溶剂挥发损失。
3.压力快速溶剂萃取
压力快速溶剂萃取(Pressurized Solvent Extraction, PSE)通过升高压力来提高溶剂的沸点,使萃取循环在常温下进行,所得到的萃取物(状态不变)维持天然活性产物原有的特性。大大缩短萃取时间提高萃取效率并显著降低溶剂消耗。此外,循环能自动重复多次,从而确保提取物的充分完全萃取,因此特别适合处理坚硬的物料如根、皮、叶等。
三、实验用品
仪器:粉碎机、样品筛、恒温水浴、超声提取仪、布氏漏斗、滤纸、真空泵、抽滤瓶、天平、圆底烧瓶、冷凝回流管、压力快速溶剂萃取仪、旋转蒸发仪等。
材料:葵花籽。
试剂:石油醚。
四、实验方法
1.葵花籽油的超声波提取法
精密称取向日葵种子100.00g,剥壳,50℃烘干,研磨,过20目筛。取15.00g粉末于150 mL锥形瓶中,加入105.00 mL石油醚。设定超声频率59 kHz,功率210W,超声时间15 min,温度40℃。三角瓶置于超声波提取仪中提取15min,重复提取两次。抽滤,滤液在旋转蒸发仪上蒸干,得到葵花籽油置于青霉素小瓶中,玻璃真空干燥器中干燥,称重。计算出油率。
2. 葵花籽油的回流提取
精密称取干燥的向日葵籽仁粉末15.00g于圆底烧瓶中,加入105.00mL石油醚(沸程30~60℃ ),回流提取6.0h,混合物经过滤后,减压旋转蒸发回收溶剂,油脂干燥后称重,计算出油率。
3. 葵花籽油的压力快速溶剂提取
精密称取120.00g葵花籽粉末,800mL石油醚,设定时间为10min,提取温度室温,压力为60bar,进行萃取。所得葵花籽油干燥后称重,计算出油率。
五、作业
1.出油率测定后测定后比较三种提取方法的优劣
实验四 高速逆流色谱法快速分离锁阳中的原儿茶酸
一、实验目的
1.了解高速逆流色谱的工作原理
2.掌握高速逆流色谱分离甘草酸的方法
二、实验原理
高速逆流色谱属于逆流色谱的范畴,它的主要分离原理是利用样品在固定相和流动相之间的差异也就是分配比不同而进行分离的,值得注意的是逆流色谱的固定相和流动相都是液体,其主要优点:(1) 无不可逆吸附。聚四氟乙烯管中的固定相无需载体液- 液色谱系统,故而消除了气- 液和固- 液色谱中因使用载体而带来的吸附现象,特别适于分离极性物质和生物活性物质;(2)高回收率。由于流动相和固定相均为液体,样品可全部回收,分离纯化与制备可同步完成,故特别适于制备性分离;(3)操作简便。因固定相为液体,体系更换与平衡方便、快捷。与HPLC 相比,HSCCC 进样量较大,最多可达数克。
三、实验用品
仪器:TBE-20A 高速逆流色谱仪、恒流泵、紫外检测器、岛津LC-20A高效液相色谱仪。
材料:干燥锁阳药材
试剂:分析纯正己烷、氯仿、乙酸乙酯、甲醇、丙酮、蒸馏水等溶剂。
四、实验方法
1.锁阳乙酸乙酯提取部位的制备
100g锁阳肉质茎粉末(过30 目筛)用300ml的70%丙酮水溶液浸提3 次,每次24 h。提取物过滤合并后40 ℃减压浓缩。粗提物用水分散后依次用正己烷(HF)、氯仿(CF)、乙酸乙酯(EAF)萃取得各萃取部分。乙酸乙酯萃取物40℃减压蒸干后用于分析与分离实验。
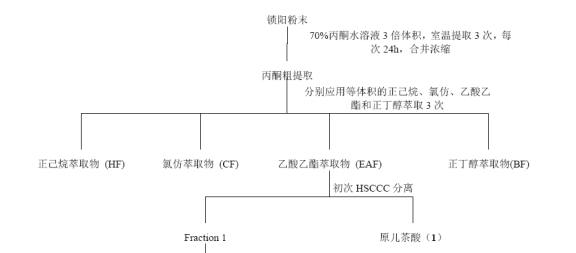
2.HSCCC溶剂体系及样品的溶解
溶剂体系:正己烷-乙酸乙酯-甲醇-水(1:3:1:3,v/v/v/v)。室温下在分液漏斗中按比例混合上述溶液,以上相作为固定相,下相作为流动相。锁阳乙酸乙酯提取物样品的溶解方案:样品溶解于20 ml 正己烷-乙酸乙酯-甲醇-水(1:3:1:3,v/v/v/v)溶液中。
3.HSCCC分离锁阳乙酸乙酯提取物
所用溶剂体系分液漏斗中配制后摇匀,过夜。分液后分别超声脱气10 min,上层液体作为固定相首先以10 ml/min 流速泵入主机直至存满柱体积,下层液体(流动相)以从头到尾的方式按2 ml/min 泵入主机,同时开启主机,转速设定为850 rpm。254 nm 处检测流出液体吸光度,约2 h 基线稳定后通过六通阀进样(清洗进样针后吸取一定量样品液后将进样针插入进样阀,切换至load状态下打入定量环,然后换回inject状态再拔针,进样后进行零点校正。inject状态下清洗进样阀)根据所得实时色谱图收集流份。
五、作业
1.记录锁阳乙酸乙酯提取物的HSCCC分离色谱图

第二篇:天然药化实验经验交流
天然药化实验经验交流
天然药物或者是中药,在我国临床上有者广泛的应用。特别是一些民间的特效药用植物,发挥着甚至是西药无法比拟的功效。近年来,国家加大了中医中药的研究投入和研究力度,这正是我们中医药工作者的良好契机,弄清楚药效物质基础是中药现代化的根本。 目前,国内外研究天然药物或者是中药的单位与研究所不计其数。研究成果也很丰硕:研究论文数量和质量逐年增加,新药开发成果也明显增多。虽然研究手段日益先进,但绝大多数的实验室或课题组仍然以传统的色谱方法或是辅以现代分析手段来提取分离天然药物或者是中药中的化学成分。我想借这个话题,给大家一个平台:分享自己的实验心得体会,共同解决探讨天然药化实验中的难题,为弄清天然药物或中药的药效物质基础贡献自己的力量。 下面,就我的实验体会谈一下我的想法。
1.在从事研究之前一定要认真查阅与课题相关的文献资料。做到心中有数,即怎样来提取是最好的?怎样来解析植物中特征类型的化合物?别人都做了哪些方面的工作了?等等。象我的课题植物主要含有生物碱类成分,我就选择先用稀酸浸泡,再用阳离子交换树脂交换富集,就把叶绿素,多糖等杂质除掉了,同时生物碱含量也提高了,给后来的分离工作带来了很大的帮助。
2.天然药物或中药一定要在做之前确定其基原(植物来源)。这很重要,不然会给文献检索带来麻烦。
3.实验过程中要粗中有细,上大柱粗分时,合并流份时要粗,就是说TLC行为大体一致的流份都可以合并,这样可以避免一些重复的实验,不然分到的成分重复的厉害。但到了后来的细分时,就要小心合并每一瓶了,不然自己的辛勤劳动就会白费了。
4.要抓重点,学会放弃。因为我觉得一个植物中的化学成分成千上百。我们不可能把它全部拿到,所以我们实验中要拿那些量稍大的、容易拿的。不要上了很久的柱子,分到
后面得到的量还不够打谱就不好了。你可以从最后结题剩下的几百个浸膏流份瓶子看到这一点。
5.实验过程中要小心每一步,三思而后行。因为你的浸膏来之不易。往往不再给你重来的机会。 6.要给自己信心,要敢于想,敢于尝试。
Sephadex LH20的分离原理主要有两方面:以凝胶过滤作用为主,兼具反相分配的作用(在反相溶剂中)。因为凝胶过滤作用,所以大分子的化合物保留弱,先被洗脱下来,小分子的化合物保留强,最后出柱。如果使用反相溶剂洗脱, Sephadex LH20对化合物还起反相分配的作用,所以极性大的化合物保留弱,先被洗脱下来,极性小的化合物保留强,后出柱。如果使用正相溶剂洗脱,这主要靠凝胶过滤作用来分离。 Sephadex LH20 洗脱溶剂。Sephadex LH20 洗脱溶剂因分为两类:反相和正相两种。用得最多的是反相溶剂洗脱,以甲醇--水系统最为常见,先用水,逐渐增加甲醇比例,最后用100%甲醇冲柱。正相系统以氯仿--甲醇最为常见,先用50%氯仿--甲醇,逐渐增加甲醇比例,最后用100%甲醇冲柱。