《水质 氨氮等6个项目的测定 气相分子吸收光谱法》
编 制 说 明
目 录
1 引言……………………………………………………………………………………… 2
2 起草单位所做的工作…………………………………………………………………… 2
3 编制标准的原则………………………………………………………………………… 3
4 标准主要内容的说明…………………………………………………………………… 3
⑴ 亚硝酸盐氮的测定…………………………………………………………………… 3
⑵ 硝酸盐氮的测定……………………………………………………………………… 6
⑶ 氨氮的测定…………………………………………………………………………… 8
⑷ 凯氏氮的测定 ……………………………………………………………………… 10
⑸ 总氮的测定 ………………………………………………………………………… 11
⑹ 硫化物的测定 ……………………………………………………………………… 12
1 引言
气相分子吸收光谱法(以下间称GPMAS)是20世纪70年代兴起的一种简便、快速的分析手段。它具有测定结果准确可靠、测定成分浓度范围宽、抗干扰性能强、不受样品颜色和混浊物的影响,不需要进行复杂的化学分离;所用化学试剂少,不使用有毒特别是易致癌的化学试剂,是一种不产生二次污染的新颖分析技术。
宝钢环境监测站始于1988年,先后研究开发出GPMAS快速测定亚硝酸盐氮和硝酸盐氮的专利方法。上报中国环境监测总站及时组织了方法验证后,国家环境保护局监督管理司于1995年4月8日发布“环监测〈1995〉079号文”,将两方法作为“水和废水监测分析方法”第三版的补充方法推广使用。之后,根据两方法原理引伸出氨氮、凯氏氮、总氮的方法,并在国内、外硫化物GPMAS的基础上,研制出更加实用的硫化物GPMAS。这一系列方法经一些分析监测单位多年应用考察和中国环境监测总站组织的方法验证及专家审定后,纳入“水和废水监测分析方法”第四版为“B”类方法。
为使这种分析技术得到更好地推广应用,宝钢环境监测站通过中国环境监测总站向国家环保总局科技标准司提出申请:“将氨氮等6个项目的气相分子吸收光谱法”列为“国家环境监测标准方法”。国家环保总局办公厅于20##年6月22日发布“环科函<20##>33号文”,授权开展方法验证工作及起草标准方法文本等。
按照国家环保局33号文件精神及中国环境监测总站的安排,由宝钢工业检测公司宝钢环境监测站负责起草《水质 氨氮等6个项目的测定 气相分子吸收光谱法》标准分析方法,组织和施实方法验证工作。宝钢环境监测站与国家环境监测总站商定协作验证单位、测试基准、水平范围,发放统一的标准样品,并负责方法验证的技术指导和对验证数据的统计分析。
参加协作验证的单位有苏州市环境监测中心站、杭州市环境监测中心站、上海市宝山区环境监测站、江苏省张家港市环境监测站、辽宁省庄河市环境监测站等5家单位。
2 标准起草单位所做的工作
2.1 进行标准分析方法的调研、查阅文献、收集资料、确定建立标准分析方法的技术路线。
2.2 将氨氮、硝酸盐氮、亚硝酸盐氮、凯氏氮、总氮及硫化物的气相分子吸收光谱法与现有国标方法进行对比分析和实验验证,确定分析结果准确可靠、具有推广应用价值的标准方法。
2.3 制订实验方案,进行分析方法研究实验,提出方法研究报告。
2.4 组织并参加方法的验证,担任协作验证实验的技术指导,对验证数据进行统计分析。
2.5 起草标准分析方法的征求意见稿及编制说明。
3 编制标准的原则
3.1 根据国家环保总局和中国环境监测总站的要求,《水质 氨氮等6个项目的测定 气相分子吸收光谱法》标准分析方法要简便快速、结果准确、具有可比性、技术先进、安全可靠、所用仪器和试剂适合我国国情,便于推广应用。
3.2 标准方法的精密度实验,按照国家《水和废水监测分析方法的标准化程序》(GB 6379—86)规定执行。
3.3 本标准分析方法,参考了1973年以来国、内外大量有关气相分子吸收光谱法的研究报告,结合我国现行国标方法进行研究制定。
4 标准主要内容的说明
4.1《亚硝酸盐氮的测定 》
4.1.1检出限及适应范围
本方法检出限是通过6个实验室各测得一批(6个)空白样的标准偏差。以3倍标准偏差除以校准曲线斜率及测定体积,得出各实验室方法检出限。然后对每个实验室得到的检出限进行统计计算,取实验室间最大值作为方法的检出限。根据6个实验室验证数据,最后得出方法的检出限为0.002mg/L(见表1)。
这一检出限低于现行方法的检出限,能满足实际监测需要。
表1 检出限测量数据
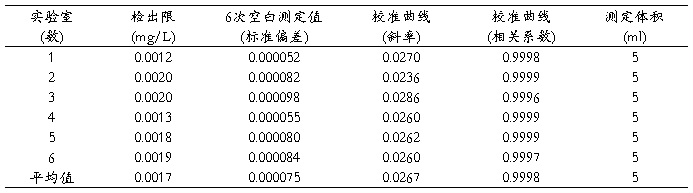
本标准方法的检出限低于N-(1-萘基)-乙二胺光度法(GB7493─87国标法),能满足实际监测需要。
当测定体积为5ml时,在锌213.9nm处测定,检测上限10mg/L。NO2气体的吸收光谱(190~300nm)呈带状,在各波长的吸收强度均与NO2-浓度成正比。在低灵敏度波长(例如锰279.5nm)处测定时,测定上限可拓宽至数百mg/L。
本标准方法适用于地表水、地下水、饮用水,特别适用于含高盐分的海水,也适用于某些生活污水和工业污水中亚硝酸盐氮的测定。
4.1.2 方法原理
酸性介质中的NO2-在乙醇等催化剂的作用下,可快速定量地分解成NO2气体,根据 NO2气体对紫外光吸收强度与NO2-浓度遵守比耳定律这一原则,利用空气为载气,将其载入测量系统,在213.9nm波长处,以校准曲线法测定亚硝酸盐氮的含量。
NO2-在酸性介质中可极其缓慢地分解生成NO2、NO、N2O3、N2O4、NOCl等多种气体,由于它们对紫外光的吸收强度极弱,没有仪器能检测出这种极弱的信号。但在酸性介质中分别加入乙醇、甲醇、甲醛等催化物质时,NO2-便迅速地全部分解成了密集的NO2气体,即使浓度远远低于mg/L级的亚硝酸盐也能容易地被检测。为了提高方法的抗干扰性能和安全性,本标准采用0.15~0.25mol/L柠檬酸及0.5ml无水乙醇为催化剂的反应介质来测定水中亚硝酸盐氮。
4.1.3 反应介质及其浓度
NO2-在HCl、H2SO4、H3PO4、柠檬酸及酒石酸等酸性介质中均可被催化剂加速分解,生成NO2气体,在同一酸度下测得的吸光度比较一致(表2)。
表2 反应介质及浓度对吸光度的影响
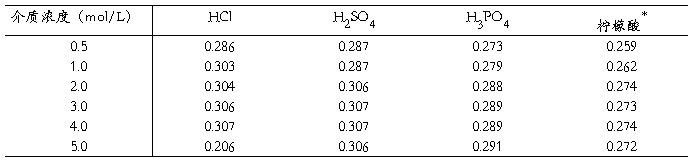
*柠檬酸浓度为0.05~0.3 mol/L。
表2说明,无机酸比有机酸测定灵敏度略高,三种无机酸的浓度从2.0~5.0mol/L时产生稳定吸收。柠檬酸浓度0.15~0.25mol/L吸光度平稳,测定灵敏度略低,但抗干扰性强,因此本标准方法采用柠檬酸介质,浓度保持在0.15~0.25mol/L之间测得的吸光度一致。
4.1.4催化剂及其用量
乙醇、甲醇、甲醛等都是很好的催化剂。甲醇及甲醛毒性大,反应时甲醇泡沫多,操作不便。因此方法采用廉价、无毒害的乙醇为催化剂。在5ml反应介质中,空白值低的乙醇其用量在0.4~0.6ml得到的吸光度稳定。对市售空白值较高的乙醇,其用量应力求准确。
4.1.5测定液体积
载气流量0.6L/min时测定液体积在4~6ml吸光度稳定。测定痕量NO2--N时,为增加取样量,也可增大体积至10ml进行测定。
4.1.6 载气及流量
以廉价的空气为载气。当测定液体积5ml时,载气流量在0.5~0.6L/min吸光度平稳。大于0.6L/min的载气流量,使出峰和回零均较快,但较低的载气流量能保持较高浓度的NO2气体,得到的吸光度较高,适合测定低含量样品。测定液体积10ml时,载气流量亦应小些。
4.1.7 催化反应时间
虽然催化剂可使NO2-瞬间分解出NO2,但测定低含量样品时,加入催化剂后约5~10s,使催化分解反应完全,得到的吸光度高且稳定性较好。
4.1.8 干扰及消除
样品中,易分解产生吸收以及能氧化或还原NO2-的物质影响测定。SO32-分解成SO2、I-、挥发成I2、S2-生成H2S,均产生吸收呈正干扰;S2O32-还原消耗NO2-,MnO4-氧化NO2-呈负干扰。
测定0.2mg/L NO2--N时,加入柠檬酸后放置1~2min,SO32-可被絡合,其量达25mg/L不影响测定;S2O32-还原NO2-不是瞬间反应,采取先加乙醇再加柠檬酸立即通气测定,允许量可达10mg/L;I2的吸收不灵敏,允许量为30mg/L;100mg/L MnO4-和80 mg/LSn2+(SnCl2)不氧化、还原NO2-;20mg/LSCN-不影片测定;大于1mg/L S2-,可以在气路中串接含乙酸铅棉的除硫管,使挥发出的H2S生成PbS而去除干扰;水样中某些产生吸收的有机物,可被活性碳吸附的,加活性碳搅拌吸附约30min,能有效去除其影响。
4.1.9 精密度和准确度
精密度:为了考查本标准方法的精密度,参加方法验证的6个单位测定了NO2--N浓度0.102mg/L±0.006mg/L的统一标样及各单位日常监测的实际样品(各重复测定6次)。经统计:重复测定的相对标准偏差为1.1%,再现测定的相对标准偏差为3.1%(表3)。
对含量为0.058~0.396mg/L的实际样品重复测定的相对标准偏差在2.3%~4.6%之间.
表3 方法的精密度和准确度

准确度:6个实验室测定NO2--N浓度0.102mg/L±0.006mg/L的统一标样,测定平均值0.102mg/L。相对误差0.0%。
对含NO2--N 0.152~2.23µg的18个实际样品进行加标回收实验,加标量为0.182~2.00µg,所得加标回收率在93.0%~106%之间(回收率93.0%的为两个)。
4.2《硝酸盐氮的测定 》
4.2.1检出限及适应范围
本方法检出限是通过6个实验室各测得一批(6个)空白样的标准偏差。以3倍标准偏差除以校准曲线斜率及测定体积,得出各实验室方法检出限。然后对每个实验室得到的检出限进行统计计算,取实验室间最大值作为方法的检出限。根据6个实验室验证的数据,最后得出方法的为检出限为0.006mg/L(见表1)。
这一检出限低于现行方法的检出限,能满足实际监测需要。
表1 检出限测量数据
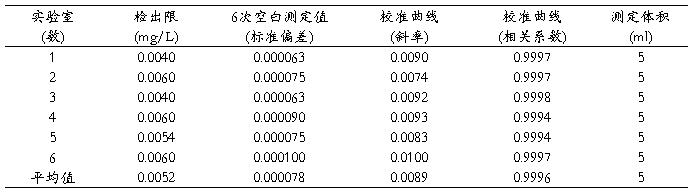
本标准适用于地表水、地下水、饮用水、海水及生活污水和工业废水中硝酸盐氮的测定。
取样2.5ml,测定体积为5ml时,在214.4nm处测定,取1ml样品,测定上限达30mg/L。
4.2.2方法原理
在HCl、H2SO4等酸性介质中,NO3-可被三氯化钛还原分解,生成的NO气体,在204.6nm、214.6nm及226.2nm处产生窄带吸收峰。204.6nm波长吸收灵敏度略高,但没有适合该波长的光源。采用吸收灵敏度稍低的214.6nm波长,找到了原子吸收用的镉空心阴极灯发射出极相近的214.4nm波长,使测定得以实现。三氯化钛还原NO3-完全生成NO气体,常温(25℃)下约需5min,在70±2℃时,可瞬间几乎完全定量地分解成NO气体。
本标准确定了在2.5~5mol/L盐酸介质中,将瞬间生成的NO气体用空气载入气相分子吸收光谱仪的测量系统,测得0~30µg NO3--N的吸光度与其浓度呈线性,以校准曲线法直接测定水样中硝酸盐氮的含量。
4.2.3 介质及其浓度
采用空白低的HCl介质。浓度在2~5mol/L,可得到稳定的吸光度。但所用HCl空白值较高时,介质浓度必须保持准确一致。
4.2.4 测定体积
测定体积在4~6ml均能得到稳定的吸光度。但反应介质空白高时,测定体积应控制一致,一般为5ml。
4.2.5 载气及其流量
生成的NO气体在短时间内不会被氧化。实验证明,用空气或氮气测得的吸光度一致性很好,因此用廉价的空气作为载气。测定体积5ml时,载气流量控制在0.5~0.6L/min,测得的吸光度高且稳定。
4.2.6 还原剂用量
100µg NO3-还原成NO气体,约需0.5ml三氯化钛。水样含氧化性物质消耗三氯化钛,应增加用量保持溶液紫红色不褪。加入量不超过2m,以免测定体积改变太大影响测定结果。
4.2.7 还原时间
在70℃±2℃可瞬间定量地还原NO3-成NO气体。加入三氯化钛溶液后,停留约10s钟,以使还原完全,可提高低含量样品的测定灵敏度。
4.2.8 干扰及消除
在HCl介质中,绝大部分阴、阳离子不被三氯化钛还原生成气体,不干扰测定。NO2-可被还原成NO2气体产生正干扰,加入氨基磺酸溶液将其完全分解生成N2气不干扰测定;SO32-及S2O32-产生正干扰,用稀H2SO4调成弱酸性,加入0.5% 高锰酸钾氧化成稳定的SO42-直至生成MnO2.H2O沉淀。取上清液测定可消除干扰;存在高价态阳离子时,应增加三氯化钛用量至溶液紫红色不褪,以保证NO3-完全被还原成NO气体;水样中含有产生吸收的有机物,可被活性碳吸附的,加活性碳搅拌吸附约30min,能有效去除其影响。
4.2.9 精密度和准确度
精密度:为了考查本标准方法的精密度,参加方法验证的6个单位测定了NO3--N浓度0.595mg/L±0.026mg/L的统一标样及各单位日常监测的实际样品(各重复测定6次)。经统计:统一标样重复测定的相对标准偏差为1.9%,再现测定的相对标准偏差为2.0%(表2)。
对含量为0.28~1.48mg/L的实际样品重复测定的相对标准偏差在1.7%~3.2%之间;
表2 方法的精密度和准确度

准确度:6个实验室测定NO3--N浓度0.595mg/L±0.026mg/L的统一标样,测定平均值0.592mg/L。相对误差1.7%。
对含NO3--N 0.763~11.75µg的18个实际样品进行加标回收实验,加标量为0.83~10.00µg,所得加标回收率在91.0%~106%之间(其中91.0%的仅有一个)。
4.3《氨氮的测定 》
4.3.1 检出限及适应范围
本方法检出限是通过6个实验室各测得一批(6个)空白样的标准偏差。以3倍标准偏差除以校准曲线斜率及测定体积,得出各实验室测得的方法检出限。然后对每个实验室得到的检出限进行统计计算,取各实验室间最大值作为方法的检出限。根据6个实验室验证的数据,最后得出方法的为检出限为0.002mg/L(见表1)。
这一检出限低于现行方法的检出限,能满足实际监测需要。
表1检出限测量数据
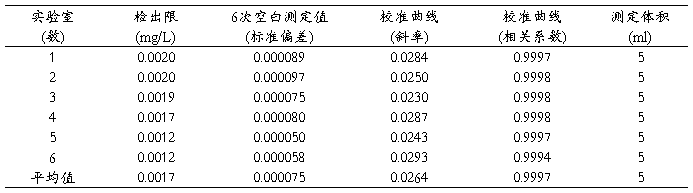
本标准适用于地表水、地下水、饮用水、海水及生活污水和工业废水中氨氮的测定。
水样中含50µg氨氮可定量地被次溴酸钠氧化为亚硝酸盐氮,取1ml样品进行测定时,测定上限可达50mg/L。
4.3.2 方法原理
水样中氨及铵盐,在碱性溶液中,经次溴酸钠氧化剂氧化成等量亚硝酸盐后,在盐酸介质中,以亚硝酸盐氮的形式测定氨氮的含量。在20%NaOH溶液中生成的次溴酸钠,氧化0~50µg氨氮成为亚硝酸盐氮的氧化率在100%~98%。必须按照这一氧化范围取样测定。
4.3.3次溴酸钠氧化剂的配制及使用
次溴酸钠氧化剂贮备液应存放在磨口玻璃瓶中,不要用塑料瓶。配制次溴酸钠使用液时,称取KBrO3和KBr的量要准确,要保证二者比例。配制使用液时,加入的氧化剂贮备液及盐酸应准确,配制用水和试剂以及室内温度应在18℃以上,可得到足够量的BrO-,达到氧化剂的氧化能力。样品加入氧化剂应充分摇动混匀,待小气泡逸尽,准确加入。在18℃以上的室温氧化时间不少于20min,以保证氨氮的氧化率。
4.3.4 测定介质及其浓度
为中和次溴酸钠氧化剂中较强的碱性,采用HCl介质。取3.0ml氧化液测定时,加入3ml 6mol/L HCl,介质酸度为2.22 mol/L,达到了测定亚硝酸盐氮的盐酸酸度(2~5mol/L)范围(见4.1亚硝酸盐氮测定的表2)。
4.3.5 测定体积、载气流量、催化剂用量及催化时间,与亚硝酸盐氮的测定一致。
4.3.6 干扰及消除
将氨及铵盐氧化成NO2-进行NH3-N的测定。含NO2-的水样,测得的氨氮包含了NO2--N,应以绘制出的同一根校准曲线,在约2~5mol/L HCl介质中快速测出NO2--N量,再测定NH3-N(包括NO2--N)的含量,差减后得到氨氮的含量。
水样中含SO32-、S2O22-、I-、SCN-等,因消耗次溴酸钠氧化剂,影响氨及铵的氧化率。当水样含0.2mg/L氨氮时,SO32-、I- 、分别小于100µg, SCN-小于50µg,或三者的混合物小于100µg,不影响测定。当氨氮在1mg/L时,上述离子的允许量仅为1/3。S2O32- 严重消耗次溴酸钠氧化剂,0.1mg/L即影响测定。个别氧化剂如 MnO4-等,在盐酸介质中可氧化转化成的NO2-,使测定结果偏低;存在能被次溴酸钠不同程度地氧化成亚硝酸盐的有机胺使测定结果偏高。若水样分别或同时含有超过上述干扰成分浓度时,必须按GB7479—87附录4蒸馏分离后,分取馏出液进行测定。氧化液的颜色和不太高的混浊物不影响测定。
4.3.7精密度和准确度
精密度:为了考查本标准方法的精密度,参加方法验证的6个单位测定了氨氮浓度1.08mg/L±0.06mg/L的统一标样及各单位日常监测的实际样品(各重复测定6次)。经统计:统一标样重复测定的相对标准偏差为1.9%,再现测定的相对标准偏差为2.5%(表2)。
对含量为0.672~2.31mg/L的实际样品重复测定的相对标准偏差在1.4%~2.7%之间;
表2 方法的精密度和准确度

准确度:6个实验室测定氨氮浓度1.08mg/L±0.06mg/L的统一标样,测定平均值1.06mg/L。相对误差1.8%。
对含氨氮 0.14~3.83µg的18个实际样品进行加标回收实验,加标量为0.1~2.0µg,所得加标回收率在93.0 %~105 %之间(回收率93.0%的仅有一个)。
4.4《凯氏氮的测定》
4.4.1检出限及适应范围
完成消解后的水样,转入100ml容量瓶中,分取10ml于50ml容量瓶中氧化成亚硝酸盐后,吸取2.5ml进行测定。检出限0.01mg/L,测定上限200mg/L。
本标准主要用于地表水、水库、湖泊和江河水中凯氏氮的测定。
4.4.2 方法原理
用硫酸加热消解水样,使游离氨、铵盐和有机物中的胺转变成硫酸氢铵,移入50ml容量瓶中,用溴百里酚蓝指示剂调至中性后,加入次溴酸盐氧化剂,将铵盐氧化成亚硝酸盐。在盐酸介质中,以亚硝酸盐氮的形式进行凯氏氮的测定。
4.4.3 样品的予处理
本方法不使用凯氏氮瓶消解样品,仅使用一般的縮口烧杯(一种小口徑的、高腰烧杯)加盖表面皿,可以保证样品消解完全。消解后的样品全部生成铵盐后,再用次溴酸钠氧化成亚硝酸盐,以亚硝酸盐氮的形式进行测定。避免了用凯氏氮瓶消解样品并进行蒸馏的烦索操作,同样使测定结果准确可靠,并能进行批量分析测定。
4.4.4 其它说明参照氨氮的测定。
4.4.5 干扰及消除
本方法样品的前处理与凯氏氮的国标法(光度法)相同。消解后样品中的阴、阳离子能氧化、还原生成的NO2-,含糊其量高时,对测定有一定的影响。
4.4.6 精密度和准确度
本方法测定的精密度和准确度取决于样品前处理,其处理方法与凯氏氮GB 11891―89国标法完全相同,测定精密度和准确度与氨氮的测定一致。
精密度实验:测定了1.00mg/L±0.05mg/L凯氏氮的统一标准样品(n=10),测得结果为0.99~1.03mg/L,平均值1.00mg/L,极差小于0.05mg/L。
在测定统一标样过程中,反复测定了近一个月时间,测定结果均是2.00mg/L。因不敢怀疑统一标样值会有问题,无耐之际,寻问了环保标样研究所,是因写错稀释倍数。纠正稀释倍数后测得的数据即是1.00mg/L。气相分子吸取光谱法测定结果的准确程度,使标样研究所有关人士及所长很感兴趣。
准确度的实验:测定1.00mg/L±0.05mg/L凯氏氮的统一标样,平均值1.01mg/L,相对误差1%;采用对多个地表水样进行加标回收试验,加入0.5~10µg凯氏氮标样,测得回收率在98.0%~101%之间。
4.5《总氮的测定》
4.5.1检出限及适应范围
在50ml容量瓶中,以碱性过硫酸钾消解水样,然后分取2.5ml进行测定。检出限0.01mg/L,测定上限100mg/L。
本标准主要用于地表水、水库、湖泊、江河水中凯氏氮的测定。
4.5.2方法原理
样品的消解完全按照已有国标法(GB 11891—89)操作。即在120~124℃碱性介质中,加入过硫酸钾氧化剂,将水样中的氨、铵盐、亚硝酸盐以及大部分有机氮化合物氧化成硝酸盐后,以硝酸盐氮的形式进行总氮的气相分子吸收光谱法测定。
4.5.3 其它说明参照硝酸盐氮的测定。
4.5.4干扰及消除
样品消解后,含高价态阳离子消耗三氯化钛还原剂,影响测定,应酌情增加三氯化钛用量,使还原后的溶液保持紫红色不褪。加入的三氯化钛不要超过1ml,以免测定体积不一致影响测定结果。
样品消解时,宜使用标线接近瓶口的容量瓶。加入样品和消解液后,加入去离子稀释至标线,减小空间,防止消解过程中挥发氨在气相损失过多,使测定结果偏低。
4.5.5精密度和准确度
精密度实验:测定总氮为3.05mg/L±0.15mg/L的标准样品(n=6),测得结果为2.95~3.04mg/L,相对标准偏差1.14%。
准确度实验: 测定总氮为3.05mg/L±0.15mg/L的标准样品,平均结果为3.00mg/L,相对误差为1.6%;对地表水样加入15.25µg总氮标样,测得回收率93.0%~101%。
4.6《硫化物的测定》
4.6.1检出限及适应范围
本方法检出限是通过6个实验室各测得一批(6个)空白样的标准偏差。以3倍标准偏差除以校准曲线斜率及测定体积,得出各实验室测得的方法检出限。然后对每个实验室得到的检出限进行统计计算,取实验室间最大值作为方法的检出限。根据6个实验室验证的数据,最后得出方法的检出限为0.003mg/L(见表1)。
这一检出限低于现行方法的检出限,能满足实际监测需要。
表1检出限测量数据
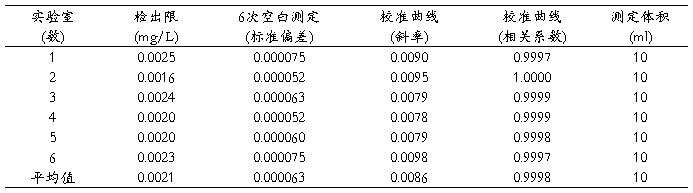
当测定体积为10ml时,在灵敏波长(如锌202.6nm)处测定,检测上限10mg/L。在低灵敏度波长(如镉228.8nm)处测定,上限可拓宽至数百mg/L。
本标准适用于地表水、地下水、饮用水、海水及各种污水中硫化物的快速测定。
4.6.2方法原理
在5%~10%磷酸介中将硫化物分解成H2S气体。将该气体用空气载入气相分子吸收光谱仪的吸光管中,在202.6 nm等波长处测得的吸光度与硫化物的浓度遵守比耳定律。以校准曲线法直接测定水样中硫化物。
H2S气体的吸收光谱(180~240nm)呈带状,在各波长测得H2S的吸收强度均与硫化物浓度成正比,因此测定浓度范围很宽(0.01~500mg/L)。
本标准仅用5%磷酸加2滴过氧化氢酸化吹气,即可快速测定基体不太复杂的水样;采用混合纤维素滤膜过滤硫化锌沉淀再酸化吹气,几乎可以测定所有水样中的硫化物。
4.6.3 测定介质及其浓度
水样中的硫化物(S2-、SH-和存在于悬浮物中的可溶性硫化物、酸可溶性金属硫化物以及未电离的有机和无机硫化物)极易被强酸(H2SO4、HCl、H3PO4)分解成H2S气体。为与已有国标法保持一致,本法采用H3PO4介质,其浓度保持在2%~10% 测得的吸光度稳定。但考虑到空白的影响,测定样品时还是应当使浓度保持一致。一般酸化吹气法浓度保持在5%;沉淀过滤─酸化吹气法,因要保证滤膜上的ZnS沉淀溶解完全,浓度宜保持在10%。
4.6.4 测定液体积
气、液相的比例对 H2S气体的挥发影响较大。在载气流量一定时,测定液体积小吸光度高,反之则低。实验证明,液相(测定液)相差0.5ml时,吸光度约差0.005Abs。因此测定样品时,应将洗净的反应瓶尽量空干一致,加入的试剂也应准确。
4.6.5 载气及其流量
实验证明,在数分钟内H2S气体被空气氧化的损失极少。本方法吹气分离测定仅10~15秒,最长不过30秒。使用廉价的空气为载气可完全满足测定的需要。载气流量的影响与其它方法相似,测定时以固定流量为好。
4.6.6 干扰及消除
在磷酸介质中NO2- 、SO32-、S2O32-产生正干扰。水样中硫化物浓度为0.5mg/L,加入2滴H2O2可分别消除1500mg/L NO2-、2000mg/L SO32-、1000mg/L S2O32的干扰;对I- 、SCN-等干扰离子及挥发性有机物的影响,采用沉淀过滤及酸化吹气的双重分离手段进行测定,对所有水样都能得到真实可靠的分析结果。
采用对ZnS无吸附的混合纤维素滤膜过滤,过滤洗涤仅3~5mi。与ZnS共沉淀的ZnSO3在H3PO4介质中生成SO2气体参与吸收,使测定结果偏高。所以溶解滤膜上的硫化物共沉淀时,须先加入2滴H2O2将ZnSO3氧化成不挥发的ZnSO4。采用沉淀过滤及酸化吹气分离手段,可以测定印染、皮革以及造纸等几乎所有污水中的硫化物。
4.6.7精密度和准确度
精密度:为了考查本标准方法的精密度,参加方法验证的6个单位测定了硫化物浓度1.97mg/L±0.09mg/L的统一标样及各单位日常监测的实际样品(各重复测定6次)。经统计:统一标样重复测定相对标准偏差为1.7%,再现测定的相对标准偏差为2.4%(表2)。
对含量为2.42~7.53mg/L的实际样品重复测定的相对标准偏差在1.4%~3.3%之间。
表2 方法的精密度和准确度

准确度:6个实验室测定硫化物浓度1.97mg/L±0.09mg/L的统一标样,测定平均值1.98mg/L。相对误差0.5%。
对含硫化物0.24~12.87µg的18个实际样品进行加标回收实验,加标量为0.50~10.00µg,所得加标回收率在92.0 %~104 %之间(回收率92.0%的只有一个)。