Realtime PCR 检测技术
一、 实验用品:
【实验耗材】
1、移液枪:1mL、100μL、10μL。酒精擦拭,头部重点,紫外线照射。
2、吸头:1mL、100μL、10μL
3、EP管:1.5mL、100μL
4、铝制饭盒
【实验仪器】
天平、振荡器、高速离心机、0.5~10uL、10~200uL、100~1000uL加样器。
【自备试剂】
DEPC(焦碳酸二乙酯),二甲苯,无水乙醇,0.1%DEPC-H2O。
实验准备:
【器具处理与准备】
先将DEPC水从容量瓶中倒入瓷缸中,将塑料制品逐个浸泡其中,小枪头需打入DEPC水,室温过夜,高压,烤干备用。
二、 样品的miRNA抽提
样品中miRNA抽提参照QIAGEN说明书(miRNeasy FFPE Handbook)
三、 RT (All-One miRNA qRT-PCR Reagent Kits,广州复能基因)
1、按下列组份配制RT反应液
5×PAP/RT Buffer 5 μL
RTase Mix 1μL
2.5U/ul Poly A Polymerase 1μL
Total RNA 18μL
Total Up to 25μL
2、反转录反应条件如下: 37℃ 60 min,85℃ 5 min。
3、反应结束后,将其放在冰上待用或-20℃保存。
四、 qPCR (All-One miRNA qRT-PCR Reagent Kits,广州复能基因)
1、按下列组份分别配制Realtime PCR反应体系,以下是每一孔20μL 。
2×All-in-One qPCR Mix 10μL
All-in-One miRNA qPCR Primer(2μM) 2μL
Universal Adaptor PCR Primer(2μM) 2μL
DNA模板(1:5稀释) 2μL
50×ROX Reference Dye 0.4μL
dH2O(灭菌蒸馏水) 3.6μL
Total 20μL
用漩涡振荡器将管中溶液彻底混合均匀,短暂低速离心。
2、点样,将步骤1中混合好的液体加入孔板中,每个样本的每个基因保证3个复孔,点完样之后将PCR板置于离心机中2000rpm、2min,然后用锡箔纸将板包好,置于4℃冰箱中备用。
引物设计

第二篇:Realtime PCR
实时荧光定量PCR:一种科学准确的定量方法
实时荧光定量PCR技术于19xx年由美国Applied Biosystems公司推出,由于该技术不仅实现了PCR从定性到定量的飞跃,而且与常规PCR相比,它具有特异性更强、有效解决PCR污染问题、自动化程度高等特点,目前已得到广泛应用。本文试就其定量原理、方法及参照问题作一介绍。
一. 实时荧光定量PCR原理
所谓实时荧光定量PCR技术,是指在PCR反应体系中加入荧光基团,利用荧光信号积累实时监测整个PCR进程,最后通过标准曲线对未知模板进行定量分析的方法。
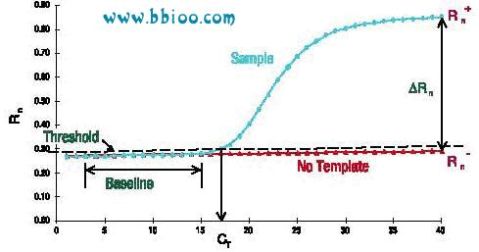
1.在荧光定量PCR技术中,有一个很重要的概念—— Ct值。C代表Cycle,t代表threshold,Ct值的含义是:每个反应管内的荧光信号到达设定的域值时所经历的循环数(如图1所示)。
图1. Ct值的确定
2.荧光域值(threshold)的设定
PCR反应的前15个循环的荧光信号作为荧光本底信号,荧光域值的缺省设置是3-15个循环的荧光信号的标准偏差的10倍,即:threshold = 10? SDcycle 6-15
3. Ct值与起始模板的关系
研究表明,每个模板的Ct值与该模板的起始拷贝数的对数存在线性关系〔1〕,起始拷贝数
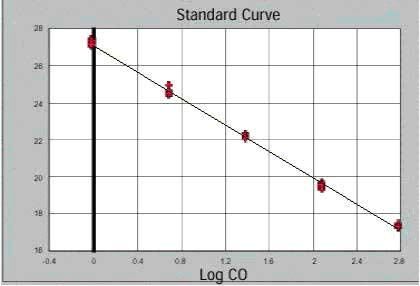
越多,Ct值越小。利用已知起始拷贝数的标准品可作出标准曲线,其中横坐标代表起始拷贝数的对数,纵坐标代表Ct值(如图2所示)。因此,只要获得未知样品的Ct值,即可从标准曲线上计算出该样品的起始拷贝数。
图2.荧光定量标准曲线
4.荧光化学
荧光定量PCR所使用的荧光化学可分为两种:荧光探针和荧光染料〔2〕。现将其原理简述如下:
1)TaqMan荧光探针:PCR扩增时在加入一对引物的同时加入一个特异性的荧光探针,该探针为一寡核苷酸,两端分别标记一个报告荧光基团和一个淬灭荧光基团。
探针完整时,报告基团发射的荧光信号被淬灭基团吸收;PCR扩增时,Taq酶的5’-3’外切酶活性将探针酶切降解,使报告荧光基团和淬灭荧光基团分离,从而荧光监测系统可接收到荧光信号,即每扩增一条DNA链,就有一个荧光分子形成,实现了荧光信号的累积与PCR产物形成完全同步。如图3所示。

图3. TaqMan荧光探针工作原理
2)SYBR荧光染料:
在PCR反应体系中,加入过量SYBR荧光染料,SYBR荧光染料特异性地掺入DNA双链后,发射荧光信号,而不掺入链中的SYBR染料分子不会发射任何荧光信号,从而保证荧光信号的增加与PCR产物的增加完全同步。
二.内标在传统定量中的意义
1.几种传统定量PCR方法简介〔3〕:
1)内参照法:
在不同的PCR反应管中加入已定量的内标和引物,内标用基因工程方法合成。上游引物用荧光标记,下游引物不标记。在模板扩增的同时,内标也被扩增。在PCR产物中,由于内标与靶模板的长度不同,二者的扩增产物可用电泳或高效液相分离开来,分别测定其荧光强度,以内标为对照定量待检测模板〔4〕。
2)竞争法:
选择由突变克隆产生的含有一个新内切位点的外源竞争性模板。在同一反应管中,待测样品与竞争模板用同一对引物同时扩增(其中一个引物为荧光标记)。扩增后用内切酶消化PCR产物,竞争性模板的产物被酶解为两个片段,而待测模板不被酶切,可通过电泳或高效液相将两种产物分开,分别测定荧光强度,根据已知模板推测未知模板的起始拷贝数〔5〕。
3)PCR-ELISA法:
利用地高辛或生物素等标记引物,扩增产物被固相板上特异的探针所结合,再加入抗地高辛或生物素酶标抗体-辣根过氧化物酶结合物,最终酶使底物显色。常规的PCR-ELISA法只是定性实验,若加入内标,作出标准曲线,也可实现定量检测目的〔6〕。
2.内标在传统定量中的作用
由于传统定量方法都是终点检测,即PCR到达平台期后进行检测,而PCR经过对数期扩增到达平台期时,检测重现性极差。同一个模板在96孔PCR仪上做96次重复实验,所得结果有很大差异(见图4),因此无法直接从终点产物量推算出起始模板量。加入内标后,可部分消除终产物定量所造成的不准确性。但即使如此,传统的定量方法也都只能算作半定量、粗略定量的方法。
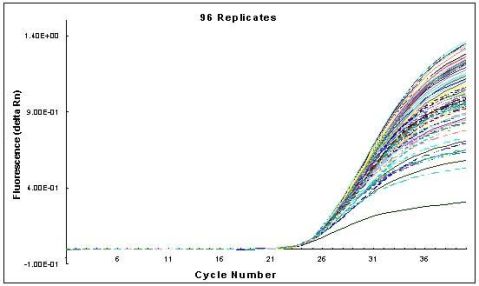
图4.相同模板在同一台PCR仪上进行96次扩增的扩增曲线图 终点处检测产物量不恒定;Ct值则极具重现性
三.内标对荧光定量PCR的影响
1.实时荧光定量PCR无需内标实时荧光定量PCR技术有效地解决了传统定量只能终点检测的局限,实现了每一轮循环均检测一次荧光信号的强度,并记录在电脑软件之中,通过对每个样品Ct值的计算,根据标准曲线获得定量结果。因此,实时荧光定量PCR无需内标是建立在两个基础之上的:
1)Ct值的重现性PCR循环在到达Ct值所在的循环数时,刚刚进入真正的指数扩增期(对数期),此时微小误差尚未放大,因此Ct值的重现性极好,即同一模板不同时间扩增或同一时间不同管内扩增,得到的Ct值是恒定的。(见图4)
2)Ct值与起始模板的线性关系由于Ct值与起始模板的对数存在线性关系,可利用标准曲线对未知样品进行定量测定,因此,实时荧光定量PCR是一种采用外标准曲线定量的方法。
2.内标对实时荧光定量PCR的影响若在待测样品中加入已知起始拷贝数的内标,则PCR反应变为双重PCR,双重PCR反应中存在两种模板之间的干扰和竞争,尤其当两种模板的
起始拷贝数相差比较大时,这种竞争会表现得更为显著〔7,8〕。但由于待测样品的起始拷贝数是未知的,所以无法加入合适数量的已知模板作为内标。也正是这个原因,传统定量方法虽然加入内标,但仍然只是一种半定量的方法。
美国Texas大学的科研人员进行了外标法定量和内标法定量的方法学比较,得出的结论是:内标法作为定量或半定量的手段是不可靠的,而外标准曲线的定量方法是一种准确的、值得信赖的科学方法〔9〕。
Applied Biosystems公司的7700型实时荧光定量PCR仪是全球公认的荧光定量PCR的金标准,可实现多色荧光同时检测,但定量检测仍然采用外标准曲线的方法,而不用内标进行定量。
综上所述,利用外标准曲线的实时荧光定量PCR是迄今为止定量最准确,重现性最好的定量方法,已得到全世界的公认,广泛用于基因表达研究、转基因研究,药物疗效考核、病原体检测等诸多领域〔10,11,12,13〕。
参考文献
1. Higuchi R, Fockler C, etal. Kinetic PCR analysis: real-time monitoring of DNA amplification reactions. Biotechnology, 1993, 11(9): 1026-1030.
2. DNA/RNA Real-Time Quantitative PCR-Rev. B Applied Biosystems
3. 定量聚合酶链反应的研究进展与临床应用.中华检验医学杂志2000, 23(2): 120-121.
4. Anderson KM, Cheung PH, Kell MD. Rapid generation of homologous internal standards and evaluation of data for quantitaion of messenger RNA by competitive polymerase chain reaction. J Pharmacol Toxicol Methods, 1997, 38: 133-140.
5. Wuhu A, Waiztu M, Koch A. A rapid and sensitive protocol for competitive
reverse transcriprase (CRT) PCR analysis of cellular genes. Brain Pathol, 1998, 8: 13-18
6. 仝文斌,高巍,费然等.核酸扩增产物的量化酶免疫通用型检测方法.中华医学检验杂志,1999, 22: 83-86.
7. Actor JK, Limor JR, Hunter RL. A flexible bioluminescent-quantitive polymerase reaction assay for analysis of competitive PCR amplicons. J Clin Lab Anal, 1999, 13(1): 40-47.
8. Schnell S, Mendoza C. Enzymological considerations for a theoretical
description of the quantitative competitive polymerase chain reaction(QC-PCR). J Theor Biol, 1997, 84(4): 433-440.
9. Ke LD, Chen Z, Yung WK. A reliability test of standard-based quantitative PCR: exogenous vs endogenous standards. Mol. Cell Probes, 2000, 14(2): 127-135.
10. Becker K., D. Pan and C.B.Whitely. Real-time quantitative polymerase chain reaction to assess gene transfer. Hum. Gene Ther, 1999, 10: 2559-2566.
11. Higgis, J.A., etal. 5’ nuclease PCR assay to detect Yersinia pesits. J Clin Microbiol, 1998, 36: 2284-2288.
12. Bieche I., P.Oondy, etal. Real-time reverse transcription-PCR assay for future management of ERBB2-based clinical apolications. Clin.Chem, 1999, 45: 1148-1156.
13. Wang X. X. Li, etal. Application of real-time polymerase chain reaction to
quantitate induced expression of interleukin-1 beta mRNA in ischemic brain tolerance. 2000, J.Neurosci.Res, 59(2): 238-46.
定量PCR Taqman探针设计要领
作者:未知 来源:pkoo.cn 时间:2007-8-1
自90年代Taqman探针诞生以来,虽然荧光探针(引物)不断有新的技术出现,但是作为一种经典的定量PCR技术,Taqman探针技术仍然是许多实验研究人员进行定量检测的首选,这主要是因为相对于SYBR荧光染料,Taqman探针具有序列特异性,只结合到互补区,而且荧光信号与扩增的拷贝数具有一一对应的关系,因此特异性强灵敏度高,而且条件优化容易;而相对于杂交探针,Taqman探针只要设计一条探针,因此探针设计较便宜方便,而且也能完成基本的定量PCR要求。当然Taqman定量方法由于还是要合成探针,也给实验操作带来了挑战。
一般Taqman定量PCR实验过程为:目的基因查找比对→探针与引物设计→探针与引物合成→配置反应体系→反应参数→重复实验,优化条件→获得曲线数据,比对标准曲线→再重复验证。
第一步:在第一步目的基因查找比对过程中可以利用NCBI genbank序列以及DNAstar等软件完成目的DNA或者RNA的查找与比对——这在分析测序报告的时候相信很多人操作过,这一步需要注意的就是要保证所分析的序列在一个contig(重叠群,即染色体的一些区域中毗邻DNA片段重叠的情况)内。
第二步:如果其它条件一致,那么这个第二步——引物探针的设计就可以说是定量PCR成败的关键了,通过各方面经验的总结有以下几个基本的原则:
总体原则
* 先选择好探针,然后设计引物使其尽可能的靠近探针。
* 所选序列应该高度特异,尽量选择具有最小二级结构的扩增片段——这是因为二级结构会影响反应效率,而且还会阻碍酶的扩增。建议先进行二级结构检测,如果不能避免二级结构,那么就要相应提高退火温度。
* 扩增长度应不超过400bp,理想的最好能在100-150bp内,扩增片段越短,有效的扩增反应就越容易获得。较短的扩增片段也容易保证分析的一致性。
* 保持GC含量在20%和80%之间,GC富含区容易产生非特异反应,从而会导致扩增效率的降低,以及出现在荧光染料分析中非特异信号。
* 为了保证效率和重复性,应避免重复的核苷酸序列,尤其是G(不能有4个连续的G) * 将引物和探针互相进行配对检测,以避免二聚体和发卡结构的形成。
引物设计原则
* 序列选取应在基因的保守区段
* 避免引物自身或与引物之间形成4个或4个以上连续配对,避免引物自身形成环状发卡结构
* 典型的引物18到24个核苷长。引物需要足够长,保证序列独特性,并降低序列存在于非目的序列位点的可能性。但是长度大于24核苷的引物并不意味着更高的特异性。较长的序列可能会与错误配对序列杂交,降低了特异性,而且比短序列杂交慢,从而降低了产量。 * Tm值在55-65℃(因为60℃核酸外切酶活性最高),GC含量在40%-60% * 引物之间的TM相差避免超过2℃
* 引物的3’端避免使用碱基A,引物的3’端避免出现3个或3个以上连续相同的碱基
* 为避免基因组的扩增,引物设计最好能跨两个外显子。
* Taqman探针技术要求片段长度在50bp-150bp
* 引物末端(最后5个核苷酸)不能有超过2个的G和C。
探针设计原则
* 探针位置尽可能地靠近上游引物
* 探针长度应在15-45bp(最好是20-30bp),以保证结合特异性
* 检测探针的DNA折叠和二级结构
* Tm值在65-70℃,通常比引物TM值高5-10℃(至少要5℃),GC含量在40%-70% * 探针的5’端应避免使用G鸟嘌呤——因为5'G会有淬灭作用,而且即使是被切割下来还会存在淬灭作用。
* 整条探针中,碱基C的含量要明显高于G的含量——G含量高会降低反应效率,这时就应选择配对的另一条链作为探针。
* 为确保引物探针的特异性,最好将设计好的序列在blast中核实一次,如果发现有非特异性互补区,建议重新设计引物探针。
Taqman MGB 探针设计
* 探针的5’端避免出现G,即使探针水解为单个碱基,与报告基团相相连的G碱基仍可淬灭基团的荧光信号。
* Tm值应为65-67℃。
* 尽量缩短Taqman MGB探针,但探针长度不少于13bp。
* 尽量避免出现重复的碱基,尤其是G碱基,应避免出现4个或4个以上的G重复出现。 * 原则上MGB探针只要有一个碱基突变,MGB探针就会检测到(MGB探针将不会与目的片段杂交,不产生荧光信号)。因此,在进行SNP检测时,为了检测到突变子,即Taqman MGB不与目的片段杂交,不产生荧光信号,探针目的片段产生荧光信号检测将探针的突变位点尽量放在中间1/3的地方。
注意:为了满足上述要求的4个条件,探针的突变位点可向3’端移动,但突变位点至少在离3’端2个碱基的前方(即必须确保探针的后两个碱基是绝对的保守),以进行SNP检测。反过来,若要进行同类检测,找的是保守片段区,探针中不应有突变位点。若探针即便是只
有13个bp,探针仍不完全保守。有几个突变,突变位点也应靠近探针的5’端,这样,即便是突变,探针也可与目的片段杂交,产生荧光信号。另一种方法是设计简并探针,也可达到即使是突变,仍可检测到突变。 第三步:寻找一家信赖的公司合成引物和探针,一般引物合成大家比较熟悉,而且价格也比较便宜(特别是这两年便宜了许多),而探针则相对来说贵了许多,一般Taqman探针合成在1000到5000元不等(不同的合成要求价钱不同)——而这只是标记价钱,序列合成基本上和引物合成价钱相似。 第四、五、六步:一般的定量PCR反应体系与普通PCR其实也差不了多少,只是要加入Taqman探针,另外不同就是分步法的不同。其中需要注意的是:
* 扩增酶最好选用热启动酶
* 引物和探针的浓度需要进行优化,有人建议从50nM开始,在50nM—900nM之间优化,一般为200nM(注意探针需要避光保存。
* 同样Mg+和酶量也需要进行优化,酶的推荐反应浓度是1.25-1.5U(50ul)
* DNA模板的添加量通常在100 ng以下,因不同种类的DNA模板中含有的靶基因的拷贝数不同,必要时可进行梯度稀释,确定最佳的DNA模板添加量。如果欲进行2 Step RT-PCR反应的第二步PCR扩增反应,第一步的RT反应液作为DNA模板时的添加量不要超过PCR反应液总体积的10%。
另外循环参数虽然在引物和探针设计完之后也就确定了,但是有时也需要进行优化。 第七步:在进行数据分析的时候,通常用不同浓度的标准样品的Ct值来产生标准曲线,然后计算相对方程式。方程式的斜
度可以用来检查PCR的效率,对于100%PCR效率来说,一个理想的斜率是3.32。最佳的标准曲线是建立在PCR的扩增效率为90%-100 %(100%意味着在每个循环之后,模板的总数将增加为前一次的2倍)的基础上。所有标准曲线的线性回归分析需要存在一个高相关系数(R2≥0.99) ,这样才能认为实验的过程和数据是可信的。使用这个方程式我们可以计算出未知样本的初始模板量。大多数定量PCR仪都有这样一个软件,它可以从标准曲线中自动地计算出未知样本的初始模板量。
这其中有两种基本的方法:绝对定量和相对定量,研究人员需要根据自己的实验目的来选择。绝对定量是指将未知样品与标准曲线相比较进行分析,一般标准品就是一个已知绝对浓度的DNA样品,要注意得是绝对定量分析的准确性是相对标准品的准确性而言的。相对定量是指两个或更多的基因互相进行比较,其结果是一个比率,没有确切的数字被检测道。
另外由于不同的样品在反应过程中存在着一定的差异,因此除了要制作标准曲线来进行定量外,还需要设计表达水平相对较为稳定的内参基因来对结果进行标准化。β-actin 和三磷酸甘油醛脱氢酶( Glyceraldehyde-3-phosphate dehydrogenase ,GAPDH) 是两种较常用的管家基因,另外还有cyclophilin,18sr RNA ,phosphoglyserokinase,beta-microglobulin,beta-glucronidase,hypoxanthine ribosyl transferase,transferring receptor 等。要注意这些基因可以被反应条件所影响,在设计定量表达研究时,保证初始对照基因的质量是必要的一步,而且严谨的研究人员会通过对一系列内参基因定量结果取几何平均数来对定量数据进行标准化。
还有一个问题就是如何判断所得到数据的好坏,关于扩增曲线,经验总结认为:
“1. 总体看曲线拐点清楚,指数期明显,扩增曲线整体平行性极好,基线平无上扬现象,低浓度样本扩增曲线指数期明显。
2. 曲线指数期斜率,反应了扩增效率,越大说明扩增效率越高。扩增效率越高,试剂灵敏度表现会越好。
3. 基线,图上的基线也就是阴性样本的扩增曲线,平直或略微下降是好试剂的表现,阴阳性清楚,不易误判。如果有上扬趋势,有可能造成阴性标本误判。
4. 曲线与曲线间平行性非常好,说明各反应管扩增效率相近,外标准定量建立在每管扩增效率一样的假定基础上,所以扩增效率越相近,定量的重复性和准确性就越好。而扩增效率相近与否,反应在扩增曲线图上就是曲线的平行性。
5. 低浓度曲线指数期明显,一方面不易出现假阴阳性误判,另一方面说明灵敏度高。”
实时荧光定量PCR实验体系的设计与优化
作者:未知 来源:基因公司 时间:2007-8-1
时荧光定量PCR以其精确、快速、方便,越来越多的应用在科研、临床及检验检疫的各个领域。但是定量PCR是对精确性要求很高的实验,不仅要求在实验前有比较完整的实验设计方案,而且实验的条件对实验结果的影响也非常大。这些都是很多老师与学生非常关心的问题,下面分别从这两个方面来对定量PCR实验做一些阐述。
定量PCR实验步骤(以mRNA为例):
1.设计实验方案:例如对样品、实验组和对照组的一个实验流程设计
2.引物和探针的设计和合成
3.抽提RNA,测定提取的RNA的浓度
4.反转录PCR
5.定量PCR
6.数据分析
在进行定量PCR实验的过程中,PCR的扩增效率是一个非常重要的影响因素,因为定量PCR原理的理论方程是基于扩增效率最大值1,因此高的扩增效率能保证定量PCR实验的精确性及重复性,影响PCR扩增效率主要有以下几个方面:1,扩增子的长度;2,扩增子的GC含量;3,扩增子、引物和探针的二级结构;4,PCR反应各组分的浓度;5,RNA或者cDNA的纯度。
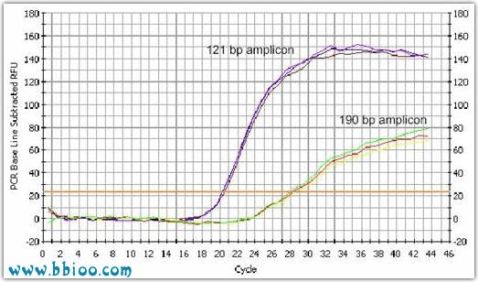
进行定量PCR实验时,必须设计好引物和探针,除了能获得高的扩增效率外,对PCR扩增的特异性、消除基因组DNA的扩增及提高扩增的灵敏度都有很大的影响。下图就是使用不同的引物和探针对18SRNA进行定量PCR的荧光曲线图(反应条件和模板都相同)。
引物设计原则:
1.上下游引物要保守为了能够扩增出所需要的保守片段,必须对保守的100-200片段进行PCR扩增。所以引物的选取也要非常的保守。
2.上下游引物的长度一般为18-30bp之间,且Tm值在58-62℃之间,上下游引物的Tm值相差最好不超过2℃。
3.确保引物中GC含量在30-80%。应避免引物中多个重复的碱基出现,尤其是要避免4个或超过4个的G碱基出现。引物的3’端最好不为G或/和C。引物3’端的5个碱基不应出现2个G或/和C。
4.避免引物内出现反向重复序列形成发夹二级结构,同时也应避免引物间配对形成引物二聚体。5.跨外显子设计引物,用于区别或消除基因组DNA的扩增。
探针设计的基本原则:
1. 保守:探针要绝对的保守,有时分型就仅仅依靠探针来决定。理论上有一个碱基不配对,
就可能检测不出来。 2. Taqman探针的长度最好在25-32bp之间,且Tm值在68-72℃之间,确保探针的Tm值要比引物的Tm值高出5-10℃,这样可保证探针在退火时先于引物与目的片段结合。
3. 确保探针中GC含量在30-80%。
4. 避免探针中多个重复的碱基出现,尤其是要避免4个或超过4个的G碱基
5. 探针的5’端不能为G,因为即使单个G碱基与FAM荧光报告基团相连时, 也可以淬灭FAM基团所发出的荧光信号,从而导致假阴性的出现。
6. Taqman探针应靠近上游引物,即Taqman探针应靠近与其在同一条链上的 上游引物。两者的距离最好是探针的5’端离上游引物的3’有一个碱基。
7. 避免探针与引物之间形成二级结构。
8. 对于多重定量PCR,例如SNP分型检测时,SNP位点应设计在探针的中间位置,并且两种探针的Tm值应相近。 实时定量PCR体系的优化
1.基本参数的优化:
1)MgCl2的浓度:在PCR反应中,MgCl2的浓度对酶的活性是至关重要的,不仅如此,合适的MgCl2的浓度还能在反应中得到较低的Cp(crossingpoint)值(指PCR达到指数扩增期时,产生一定的荧光高于背景并为仪器所识别时的循环数),较高的荧光信号强度以及良好的曲线峰值。所以对其的浓度选择应慎重。一般来说,对以DNA或cDNA为模板的PCR反应,应选择2-5mM浓度的MgCl2,对以mRNA为模板的RT-PCR而言,则应选择的浓度为4-8mM。
2)模板的浓度:如果研究者是进行首次实验,那么应选择一系列稀释浓度的模板来进行实验,以选择出最为合适的模板浓度,如果条件困难,也至少要选择两个稀释度(高和中、低浓度)来进行实验。一般而言,使Cp位于15-30个循环比较合适,若大于30则应使用较高的模板浓度,如果Cp小于15则应选择较低的模板深度。对于Cp值的确定,经验上是SYBRGreenI探针的荧光信号比本底高2倍,杂交探针的荧光强度比本底高0.3倍。
2.使用SYBRGreenI测定DNA时的条件优化:
1)MgCl2的浓度:大多数引物-模板对其的要求是2-4mM。
2)模板的浓度:初次实验要求做一系列的稀释浓度,如条件限制,至少完成两个稀释的度
的测定。基因组DNA在50ng-5pg之间选择,质粒DNA在106拷贝数左右。
3)引物的浓度:引物的浓度是一个影响PCR反应的关键因素,若浓度太低,会致使反应不完全,若引物太多,则发生错配以及产生非特异的产物的可能性会大大增加。对于大多数PCR反应,0.3uM是个合适的浓度,若初次选用这个浓度不理想,可在0.1-1.0uM之间进行选择,直至达到满意的结果。
4)退火温度:首次实验设置的退火温度应比计算得出的Tm值小5℃,然后在1-2℃内进行选择。一般的,退火温度要根据经验来确定,这个经验值往往会同计算得到的Tm值有一定的差距。
3.用SYBRGreenI进行一步法RT-PCR的条件优化:
1)MgCl2的浓度:不同的靶分子选用不同的浓度,通常是在4-8mM之间选择。
2)模板的浓度:RT-PCR实验既可以选用总RNA,又可以选用mRNA,其浓度应在1pg-1ug之间选择。对于低模板浓度,可以增加适量的MS2或用alternativeRNA作为载体进行测定。
3)对照设置:每一引物都应设有阴性对照,阳性对照和污染对照。
4.杂交探针测定DNA
1)MgCl2的浓度:在2-4mM的基础上加0.5-1.0mM,但是不要超过2.0mM。
2)杂交探针的浓度:初次实验每个探针用0.2uM,如果信号强度达不到要求,可以增加至0.4uM。
3)对照设置:每一引物都要设阴性对照,每一探针都要设阴性对照。每次实验都要设阳性对照。
4)其它的条件同SYBRGreenI。
5.用杂交探针进行实时定量RT-PCR:
1)MgCl2的浓度:在4-8mM之间进行选择。
2)杂交探针的浓度:初实验用0.2uM,如果荧光信号强度不足,可以增加至0.4uM。
3)模板浓度设置:优化的扩增须进行一系列稀释度的实验,在条件有困难的情况下,至少要进行两个稀释度的测定。选用1pg-1ug的总RNA或是mRNA,若是模板的浓度过小(小于10ng/ul),则可加入MS2或alternativeRNA作为载体。
4)对照设置:每个引物都要设无模板对照,阳性对照以及污染对照。
6.关于杂交探针的评价:在使用杂交探针进行实验时,必须注意防止探针-引物二聚体的形成和其本身在反应过程中的延伸。引物-探针二聚体的形成,主要是因为探针可与引物的3’末端杂交,其形成以后,会致使此二聚体扩增,从而同目的基因竞争反应的原料,导致反应
的效率下降。探针其本身能同目的基因相结合,且其解链温度高于引物,所以它可能作为引物而引发延伸反应,为了防止发生这种现象,通常是将其3’末端完全磷酸化,使之不能延伸,若此磷酸化不完全或是没有磷酸化,就会产生目的基因的副产品,从而干扰实验结果。鉴于以上这两点,所以应对探针精心设计,并将其末端完全磷酸化。
定量PCR常见问题及对策
作者:未知 来源:基因公司 时间:2007-8-1
Q1.无CT值(信号)出现
A1.1.反应循环数不够。一般都要在35个循环以上,可根据实验情况增加循环(如至45cycles),但高于45个循环会增加过多的背景信号。
2.检测荧光信号的步骤有误。一般SG法采用72℃延伸时采集,Taqman法则一般在退火结束时或延伸结束采集信号。
3.引物或探针降解。可通过PAGE电泳检测其完整性。
4.引物或探针的设计,如探针高于引物的温度不够,造成探针未杂交上而产物已延伸的情况。
5.模板量不足。对未知浓度的样品应从系列稀释样本的最高浓度做起。
6.模板降解。避免样品制备中杂质的引入及反复冻融的情况。
Q2.CT值出现过晚
A2.1.扩增效率低,反应条件不够优化。设计更好的引物或探针;改用三步法进行反应;适当降低退火温度;增加镁离子浓度等。
2.PCR各种反应成分的降解或加样量的不足。
3.PCR产物太长。一般采用80-150bp的产物长度。
Q3.标准曲线的线性关系不佳
A3.1.加样存在误差,使得标准品不呈梯度。
2.标准品出现降解。应避免标准品反复冻融,或重新制备并稀释标准品。
3.引物或探针不佳。重新设计更好的引物和探针
4.模板中存在抑制物,或模板浓度过高
Q4.阴性对照也出现明显的起飞
A4.1.应mix或水被污染。
2.引物二聚体的出现。用SG法在35cycles以后阴性出现起飞属正常情况,可配合熔解曲线进行分析。
3.反应过程中探针的降解。用PAGE电泳对探针进行检测。
4.如果使用了ROX校正,则可能是ROX的降解所造成。
Q5.在溶解曲线前是否插入一个同溶解程序初始温度相同的一个保温过程
A5.如果在熔解曲线前没有一个保温过程,则曲线会在前几个循环非常陡峭,使真正的熔解峰型几乎看不到。
Q6.熔解曲线不止一个主峰
A6.1.引物设计不够优化。应避免引物二聚体和发夹结构的出现。
2.引物浓度不佳。适当降低引物的浓度,并注意上下游引物的浓度配比。
3.镁离子浓度过高。适当降低镁离子浓度,或选择更合适的mix试剂盒。
4.模板有基因组的污染。RNA提取过程中避免基因组DNA的引入,或通过引物设计避免非特异扩增。
Q7.扩增效率低
A7.1.反应试剂中部分成分特别是荧光染料降解。
2.反应条件不够优化,可适当降低退火温度或改为三步扩增法。
3.反应体系中有PCR反应抑制物。一般是加入模板时所引入,应先把模板适度稀释,再加入反应体系中,减少抑制物的影响。
Q8.实验重复性不好
A8.1.加样不准确。
2.仪器在样品上温度条件有差异,即温度均一性不好。
3.模板浓度低。样品初始浓度越低,重复性越差,应减少样品的稀释倍数。
Q9.变性温度是否合适
A9.大部分的双链DNA在95°C就开始解链了,在有些情况下在90至94°C都不能很好地变性DNA。由于部分变性,参加反应的探针和引物相应的减少了,因此反应效率会下降。
Q10.变性时间是否合适
A10.15到20秒对于变性扩增子是足够了,然而,长一点的产物,可能会需要30秒,60秒的变性时间通常是不需要的。
Q11.退火温度和时间是否合适
A11.检查引物和探针的Tm值,SYBRGreenI的退火时间大约20到35秒,双标记探针通常是两步法,退火和延伸合并为一步,时间大约是45到60秒,温度通常在60°C,对于FRET探针退火步骤大约20到30秒。
Q12.在哪一步骤采集信号
A12.SYBRGreenI应当在72°C,此时绝大部分的DNA是双链状态,如上所述,双标记探针通常是两步检测,因此信号采集应该在退火延伸的整合步骤。对于FRET检测,数据应该在退火步骤检测。如果对于信号采集点不确定,可以多点采集作对比。如果监控屏幕检测不到信号,检查程序设定是否存在至少一个的信号采集点。
Q13.是否选定了合适的增益值
A13.一些情况下会看到曲线超过了窗口范围,在荧光强度100的地方成一直线。尽管大部分的数据可以用,但是减少增益,一些原始数据就不会超出范围。有时,在第一个循环信号就跳出了窗口范围,这是由于增益选得太高,看起来像没有检测到数据。如果熔解曲线分析时,开始荧光就达到了100,则应当用低的增益重新运行,而不需要重新运行扩增反应,SYBRGreenI和FRET的样品可以在一定程度上反复使用
实时定量PCR完全手册
作者:基因沉默 来源:基因沉默GOOGLE讨论组 时间:2007-8-11
方法简介
所谓的实时荧光定量 PCR 就是 通过对 PCR 扩增反应中每一个循环产物荧光信号的实时检测从而实现对起始模板定量及定性的分析。在实时荧光定量 PCR 反应中,引入了一种荧光化学物质,随着 PCR 反应的进行,PCR 反应产物不断累计,荧光信号强度也等比例增加。每经过一个循环,收集一个荧光强度信号,这样我们就可以通过荧光强度变化监测产物量的变化,从而得到一条荧光扩增曲线。
RT-qPCR是由三个步骤组成:
1.反转录:依赖反转录酶将RNA反转录成cDNDA;
2.扩增:用PCR的方法扩增cDNA;
3.检测:实时检测和定量扩增的产物.
RT-qRCR影响分析可靠性关键点(Key porint):
1.分析结果依赖于模板的数量、质量以及合理的检测方法设计
2.反转录反应的非标准化影响试验的稳定性
3.数据分析应该高度客观,如果不合理的分析,从分析结果中会得到混淆的错误结果,因此通过对RT-qPCR的每一组分进行质量评价以达到最小化变异性,最大化可重复性,而且还需要沿用一个通用的数据分析的指南。对基因表达分析的标准化的需要是与人类临床诊断分析相适应的。

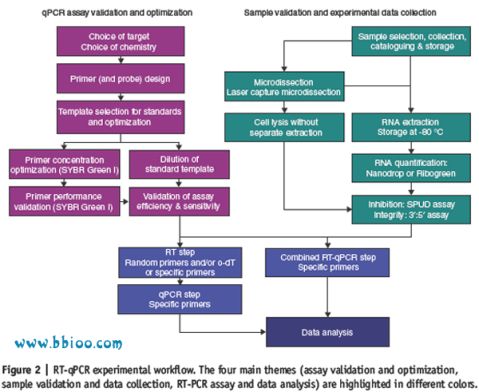
存在的问题
由于各个学术团体和科研机构使用不同的操作流程,必然导致大家使用不同定量的来源物以及数据分析:
1.新鲜、冰冻、甲醛固定的样品
2.整个组织样本,显微切割样本,单个细胞,组培细胞
3.总RNA或者mRNA
4.RNA反转录成cDNA的不同的引发策略
5.不同的酶以及酶的不同组合
6.变异系数、灵敏度
7.多类型的检测化学方法,反应的条件,热循环仪的分析以及汇报方式。
8.每一步骤缺乏标准化分析流程造成了在样品的处理,内参的使用,归一化的方法,质量控制等等因素严重影响RT-qPCR的可信度,重复性。

RNA 质量评价
现在RNA 定量的程序很多。最近EMBO qPCR course
(http://www-db.embl.de/jss/EmblGroupsOrg/conf_28) 比较了用Ribogreen, Agilent
BioAnalyser, spectrophotometer,Nanodrop and the BioRad Experion 来定量同样的样品。结果显示没有哪两种方法得到同样的分析数据。所以用不同的方法进行定量是不明智的。因此,我们需要用统一套定量分析方法来完成所有RNA样品的评价。
RNA 质量
RNA 质量主要包括RNA的纯度(没有蛋白质和DNA的污染)以及完整性。传统的RNA质量的评价通过分析A260/A280的比值或者对琼脂糖凝胶电泳rRNA的条带的分析。Agilent Bioanalyser/BioRad Experion 微流体毛细电泳系统也是一种较新的分析方法。Agilent的2100也是一种十分好的分析RNA质量的方法,它通过分析18S以及28S rRNA的分析图谱,通过图谱来反应RNA的量和完整性,其完整性通过完整性系数(RIN)来反应。样品的RINs在10-4之间。10代表完整的RNA,4代表没有完整的rRNA带。
由于以上的方法并非100%准确定反应mRNA的完整性,因为他们只是反应rRNA的量来间接测定mRNA的完整性。这里推荐一种方法:采用GAPDH的3’:5’分析法。
我们使用oligo dT进行逆转录,然后对逆转录的cDNA用multiplex荧光定量评价。设计三个taqman探针来定量三种相同大小的扩增产物。探针设计的位点分别位于3’;5’以及中部。扩增产物的之间的比值反应RNA的完整性。如果3’;5’的比值在1,反应较高度完整性,如果高于5说明降解。
QRT-PCR 抑制物的组成
QRT-PCR抑制物严重减少了PCR的灵敏度以及热动力学反应,高度的抑制还导致假阴性的结果。
抑制物的来源:生物样品的核酸抽提以及共沉淀中的混合物,盐离子,尿素,血红素,heparin以及IgG.
是否有抑制物的评价体系:
1.通过对目的样品进行梯度稀释进行PCR扩增效率的检测
2.通过内部扩增对照来反应样品处理过程中样本的情况
3.用细菌基因组检测临床样品的抑制
4.通过标准人工合成的扩增进行RT-PCR来反应目标检测物的抑制情况
反转录反应系统
1.RT和PCR单一酶系统
2.RT和PCR分离的酶系统
3.RNA逆转录引物的选择
引物主要有三种:
1.随机引物:随机引物,特别是6nt引物对所有的靶位点不产生十分稳定一致的结果,建议使用15nt的随机引物.
2.oligo-dT:只能用于mRNA完整的样品,特别有polyA .而且对于一些特殊的变异体以及较长的3’UTR的区域比较困难
3.特异引物:最特异最灵敏的方法。特别RNA量足够情况下建议使用此法。
PCR 优化
PCR优化主要有:
1.引物的浓度
2.建议使用SYBR Green I和EvaGreen 进行扩增和溶解曲线的测试
笔者建议的操作流程:
I.靶的选择和试验设计
1.针对目的基因序列选择合适的扩增片断
查看以下三个网站是否有合适的已经证实的QRT-PCR的扩增引物,探针以及反应条件. RTPrimerDB (http://medgen.ugent.be/rtprimerdb),
PrimerBank (http://pga.mgh.harvard.edu/primerbank/index.html)
Real Time PCR Primer Sets ()
如果没有合适的或者已经证实的可以提供参考,以下的设计方案仅供参考:
A.最广泛使用的商业化的软件Beacon Designer()
B.DIY的软件Primer Express
C.如果Beacon Designer 无法得到您说需要的结果,或者获得到设计方案的备选数目不够的话,可以选择Sigma-Genosys )的服务方案,详细周到
D.一个免费的基于网页构架的引物和探针设计程序:
/products/probe_design.asp
您可以挑选其中的4-6对引物进行试验,选择引物的扩增效率和灵敏度高的.这个软件可以直接与NCBI的网站进行比对,并且用NCBI的ePCR进行虚拟的电子PCR
引物设计简介
DNA引物长度:15-25 个碱基
GC含量:50%左右
如果引物的与AT区域富集结合,可以考虑用LNA替换几个碱基,较少引物的长度以及避免引物次级结构和3’端二聚体的影响.由于引物和模板和探针与靶点之间的分之间的相互竞争,分之内杂交,倒转重复等等会引起引物的引发探针对结合效率达降低,因此我们选择引物二聚体的△G为负值,即:<10 kcal/mol.没有连续的G/C.
引物探针的保存一般遵循以下原则:
正向和反向引物保存在-20度, 浓度为10mM 或者10×工作浓度.探针应该避光保存,贮存在-70度,最好以冻干粉状态,工作浓度的液体保存一般两周左右。
2.输入靶序列,用BLASTn在http://www.ncbi.nlm.nih.gov/blast进行比对
3.检查比对序列的多态性以及可能的错误避免这些区域来进行引物和探针设计
4.在靶序列中避免直接待重复区,在重复区进行杂交容易使得引物非获得产物性结合,降低DNA的扩增效率以及减少分析的灵敏度。
5.考虑到潜在的剪接变异体以及合适的所需要的获得到靶,通过生物信息学分析内含子以及外显子的边界,主要通过cDNA和基因组序列比对来确定。一般都设计跨最长内含子区,这样减少了扩增子受到基因组DNA的污染的影响。这是十分有必要的,特别当用基因组DNA做归一化处理以及靶向某一特异的剪接变异体。最经济的做法是让下游引物跨越剪接接头,这样允许使用一条探针检测可能剪接变异体.然后如果在有效性和灵敏度无法保证情况下,可以使用跨越单一外显子的设计方案。我们还是建议试验者用DnaseI处理样品,除去gDNA的污染。
6.在RT步骤时,用(http://www.bioinfo.rpi.edu/applications/mfold/rna/form1.cgi)工具检测在特定温度下靶序列的折叠情况,避免一些高度次级结构的区域,那些区域探针和引物结合效率较低
7.尽可能用60-150bp的扩增产物,GC含量在60%或者稍小来确定高效的变性,更高度反应效率。GC含量高度序列容易产生非特异性的反应,短序列扩增是
的扩增时间缩短gDNA污染可能性减少。短的序列容易人工合成,用来做扩增多标准曲线。用oligodT进行逆转录最好设计扩增子位于靠近模板的3’区域