实验二 有机物沸点及折光率的测定(4课时)
一、实验目的
1、熟悉蒸馏和测定沸点的原理,了解蒸馏和测定沸点的意义;
2、掌握蒸馏和测定沸点的操作要领和方法。
二、实验原理
沸点 当液体加热时,有大量的蒸汽产生,当内部饱和蒸汽压与外界施加给液体表面的总压力(通常为一个大气压力)相等液体开始沸腾,此时的温度为该液体化合物的沸点。 不同的化合物由于内部饱和蒸汽压达到一个大气压时的温度不同,因此沸点不同。
蒸馏 就是利用了这个特点将液体混合物加热至沸腾,使液体汽化,由于混合物中各组份的沸点不同。因此,在低沸点时蒸汽的组成以低沸点化合物为主,在相对较高沸点时蒸汽的组成以高沸点化合物为主。我们通过冷凝的方法收集不同沸点时的蒸汽,可以将混合物完全分离成单一组分。
蒸馏原理 蒸馏就是将液体混合物加热至沸腾,使液体汽化,然后,让蒸汽通过冷凝的方法变为液体,通过收集不同沸点下的蒸汽冷凝液,使液体混合物分离的过程,从而达到提纯的目的。
馏头 在达到欲收集物的沸点之前,常 有沸点较低的液体流出,这部分馏出液称为馏头或前馏分蒸 馏
蒸馏的用途 蒸馏是分离和提纯液态有机化合物的最常用的重要方法之一,还可以用来测定物质的沸点和定性地检验物质地纯度。一般来说,在合成完成后,先用简单蒸馏将低沸点的溶剂去除,然后再用其它方法进一步将化合物提纯。
三、药品和仪器
药品 : 工业 乙醇
仪器: 蒸馏瓶 温度计 直型冷凝管 尾接管 锥形瓶 量筒
四、实验装置:
搭装: 从下向上,从左到右
拆卸:正好相反)
温度计的位置:温度计水银球的上限与蒸馏头支管的下限相切。
蒸馏时必须加入沸石2~3粒,用后不许倒入水槽,以免堵塞下水管
冷却水的正确连接方式,下进上出。
选择仪器大小的标准:样品总体积不得超过烧瓶体积得2/3
沸石的作用:防止液体暴沸,使沸腾保持平稳。当液体加热至沸点时,沸石的空隙能产生很多细小的气泡,形成沸腾中心,在持续沸腾时,沸石可以继续有效。一旦停止沸腾或中途停止蒸馏,则原有的沸石即失效,应补加新的沸石。
注意:决不能在液体加热近沸腾时补加沸石,否则会引起暴沸,使液体冲出瓶外,发生着火事故。
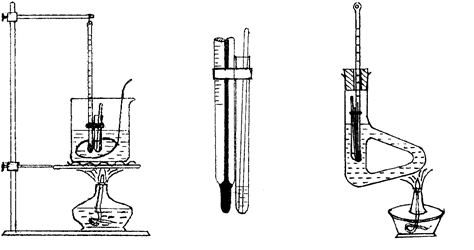
主要由气化、冷凝和接收三部分组成 ,如图4-7所示:
1、蒸馏瓶:蒸馏瓶的选用与被蒸液体量的多少有关,通常装入液体的体积应为蒸馏瓶容积1/3-2/3。液体量过多或过少都不宜。(为什么)?在蒸馏低沸点液体时,选用长颈蒸馏瓶;而蒸馏高沸点液体时,选用短颈蒸馏瓶。
2、温度计:温度计应根据被蒸馏液体的沸点来选,低于100℃,可选用100℃温度计;高于100℃,应选用250-300℃水银温度计。
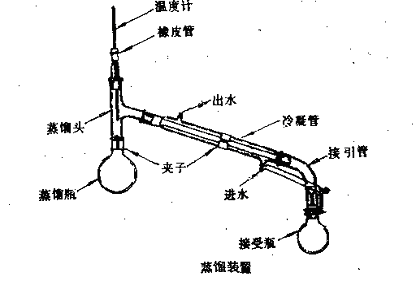
图4-7 蒸馏装置
3、冷凝管:冷凝管可分为水冷凝管和空气冷凝管两类,水冷凝管用于被蒸液体沸点低于140 oC;空气冷凝管用于被蒸液体沸点高于140 oC(为什么)。
4、尾接管及接收瓶:尾接管将冷凝液导入接收瓶中。常压蒸馏选用锥形瓶为接收瓶,减压蒸馏选用圆底烧瓶为接收瓶。
仪器安装顺序为:先下后上,先左后右。卸仪器与其顺序相反。
五、实验步骤
1、加料:将待蒸乙醇30ml小心倒入50ml蒸馏瓶中,不要使液体从支管流出。加入几粒沸石(为什么),塞好带温度计的塞子,注意温度计的位置。再检查一次装置是否稳妥与严密。
2、加热:先打开冷凝水龙头,缓缓通入冷水,然后开始加热。注意冷水自下而上,蒸汽自上而下,两者逆流冷却效果好。当液体沸腾,蒸气到达水银球部位时,温度计读数急剧上升,调节热源,让水银球上液滴和蒸气温度达到平衡,使蒸馏速度以每秒1—2滴为宜。此时温度计读数就是馏出液的沸点。
蒸馏时若热源温度太高,使蒸气成为过热蒸气,造成温度计所显示的沸点偏高;若热源温度太低,馏出物蒸气不能充分浸润温度计水银球,造成温度计读得的沸点偏低或不规则。
3、收集馏液:准备两个接受瓶,一个接受前馏分或称馏头,另一个(需称重)接受所需馏分,并记下该馏分的沸程:即该馏分的第一滴和最后一滴时温度计的读数。
在所需馏分蒸出后,温度计读数会突然下降。此时应停止蒸馏。即使杂质很少,也不要蒸干,以免蒸馏瓶破裂及发生其它意外事故。
4、拆除蒸馏装置:蒸馏完毕,先应撤出热源,然后停止通水,最后拆除蒸馏装置(与安装顺序相反)。
六、实验注意事项
1、冷却水流速以能保证蒸汽充分冷凝为宜,通常只需保持缓缓水流即可。
2、蒸馏有机溶剂均应用小口接收器,如锥形瓶。
3、工业乙醇因来源和制造厂家的不同,其组成不尽相同,其主要成分为乙醇和水,除此之外一般含有少量低沸点杂质和高沸点杂质,还可能溶解有少量固体杂质。通过简单蒸馏可以将低沸物、高沸物及固体杂质除去,但水可与乙醇形成共沸物,故不能将水和乙醇完全分开。蒸馏所得的是含乙醇95.6%和水4.4%的混合物,相当于市售的95%乙醇。
七、思考题
1、什么叫沸点?液体的沸点和大气压有什么关系?文献里记载的某物质的沸点是否即为你们那里的沸点温度?
2、蒸馏时加入沸石的作用是什么?如果蒸馏前忘记加沸石,能否立即将沸石加至将近沸腾的液体中?当重新蒸馏时,用过的沸石能否继续使用?
3、为什么蒸馏时最好控制馏出液的速度为1-2滴/S为宜?
4、如果液体具有恒定的沸点,那么能否认为它是单纯物质?
液体有机化合物折光率的测定
一、实验目的
折光率是有机化合物最重要的物理常数之一.作为液体物质纯度的标准,它比沸点更为可靠。利用折光率,可以鉴定未知化合物,也用于确定液体混合物的组成。
物质的折光率不但与它的结构和光线有关,而且也受温度、压力等因素的影响。所以折光率的表示,须注明所用的光线和测定时的温度,常用ntD表示。
通过实验让学生
1、了解阿贝折光仪的构造和折光率测定的基本原理。
2、掌握测定液态有机化合物折光率的方法。
二、基本原理
一般地说,光在两个不同介质中的传播速度是不相同的,所以光线从一个介质进入另一个介质,当它的传播
方向与两个介质的界面不垂直时,则在介面处的传播方向发生改变,这种现象称为光的折射现象。
测定:用丙酮清洗镜面后,滴加1-2滴样品于毛玻璃面上,闭合两棱镜,旋紧锁钮。调节反射镜使光进入棱镜组,并从测量望远镜中观察,使视场最明亮,再调节目镜,使视场十字线交点最清晰。再次转动罩外手柄,使刻度盘上的读数逐渐增大,直到观察到视场中出现的半明半暗现象,并在交界处有彩色光带,这时转动消色散手柄,使彩色光带消失,得到清晰的明暗界线,继续转动罩外手柄使明暗界线正好与目镜中的十字线交点重合。从刻度盘上直接读取折光率。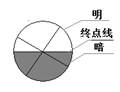
五、注意事项
1、要特别注意保护棱镜镜面,滴加液体时防止滴管口划镜面,滴管的末端切不可触及棱镜。
2、每次擦拭镜面时,只许用擦镜头纸轻擦,测试完毕,也要用丙酮洗净镜面,待干燥后才能合拢棱镜。
3、不能测量带有酸性、碱性或腐蚀性的液体。
4、测量完毕,拆下连接恒温槽的胶皮管,棱镜夹套内的水要排尽。
六、思考题
1、为什么蒸馏不能密封,应通大气?
2、为什么蒸馏不能将液体蒸干要留有一定量的残液?
3、蒸馏中,温度计水银球有无液滴意味着什么?
4、蒸馏时加入沸石的作用是什么?如果蒸馏前忘记加沸石,能否立即将沸石加至将近沸腾的液体中?当重新蒸馏时,用过的沸石能否继续使用?
5、蒸馏时最好为什么控制馏出液的速度为1-2滴/S为宜?
第二篇:实验一有机化合物熔点和沸点的测定
实验一有机化合物熔点和沸点的测定
一、有机化合物熔点的测定:
(一)实验目的
1.了解有机化合物熔点、沸点的概念、测定的原理及意义。
2.掌握微量法测定熔点、沸点的操作技术。
物质熔点的测定是有机化学工作者经常用的一种技术,所得的数据可用来鉴定晶状的有机化合物,并作为该化合物纯度的一种指标。
测定的意义:可以鉴别未知的固态化合物和判断化合物的纯度。
(二)熔点测定原理
什么叫熔点——用物质的蒸气压与温度的关系理解。熔点的定义:固、液两态在标准大气压下达到平衡状态,即固相蒸气压与液相蒸气压相等时的温度。固态物质受热后,从开始熔化(初熔)至完全熔化(全熔)的温度范围就是该化合物的熔点(实际上是熔点范围。称为熔程或熔距。)
测熔点时几个概念:始熔(初熔)、全熔、熔点距、物质纯度与熔点距关系。
始熔(初熔)——密切注意熔点管中样品变化情况。当样品开始塌落,并有液相产生时(部分透明),表示开始熔化(初熔),即记录为初溶温度t1。
全熔——当固体刚好完全消失时(全部透明),则表示完全熔化(全熔)。记录温度t2 。
熔距或熔程——从初熔到全熔的温度范围。t1~t2为熔程。纯净物一般不超过0.5~10C
化合物的熔点是指在常压下该物质的固—液两相达到平衡时的温度。但通常把晶体物质受热后由固态转化为液态时的温度作为该化合物的熔点。纯净的固体有机化合物一般都有固定的熔点。在一定的外压下,固液两态之间的变化是非常敏锐的,自初熔至全熔(称为熔程) 纯净的固体有机化合物转化为液态时的温度不超过0.5-1℃。若混有杂质则熔点有明确变化,不但熔点距扩大,而且熔点也往往下降。因此,熔点是晶体化合物纯度的重要标志。有机化合物熔点一般不超过350℃,较易测定,故可借测定熔点来鉴别未知有机物和判断有机物的纯度。
(三)熔点测定方法:
1)显微熔点测定仪 《实验化学》第二版书上P104
2)数字熔点测定仪 《实验化学》第二版书上P105
3)双浴式熔点测定器 《实验化学》第二版书上P102
4)毛细管法测熔点,用b形管测熔点装置(本实验使用)及其它测定方法。
(四)实验仪器及药品
毛细管法测熔点,用b形管测熔点装置(本实验使用)
(1)实验仪器:b形熔点测定管、玻璃管(30—40cm)、温度计、酒精灯、表面皿、毛细熔点管、指形管(沸点管)
(2)药品:苯甲酸、尿素、苯甲酸加尿素混合物、酒精、
测定物导热液为甘油——测定完毕需回收,请倒回原瓶中
测定熔点装置图:
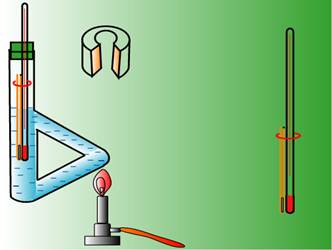

(五)实验步骤及实验关键
1.样品填装——将0.1~0.2克待测样品粉末(干燥、研碎迅速)放在干净的表面皿上聚成小堆,将毛细管开口垂直插入此小堆内将样品挤入毛细管中,在桌面上踮几下,再在玻璃管中自由落下十次左右,使样品填装结实、均匀、紧密,高度2~3mm为宜。(因测定时到了初熔时样品塌落下来,如果中间有空隙,会误认为样品塌落,造成测定偏差。)
2.毛细管安装在与温度计用橡皮圈捆在一起,橡皮圈要在导热液面上,样品位于水银球的中部,导热液在b形管的上叉管处,导热液不宜加多,以免受热后膨胀溢出引起危险,另外避免毛细管飘浮,影响准确性。再通过开口像皮塞固定在b形管中心位置,温度计与样品毛细管位于b形管上下叉管中间,此处对流循环好,温度均匀。请见测定熔点装置图:
3.小心加热b形管侧管底部,升温测定、加热升温速度:对于已知样品开始时升温可快些5~6℃/min,将近熔点10℃时,升温1~2℃/min,接近熔点时升温0.2~0.3℃/min,密切注意熔点管中样品变化情况。当样品开始塌落,并有液相产生时(部分透明),表示开始熔化(初熔),(此时应随时停止加热)当固体刚好完全消失时(全部透明),则表示完全熔化(全熔)。注意观察、做好记录。写出初熔t1~全熔t2及熔程。对于未知物需粗测一次,再精测二次。精测时方法照上述所进行。
毛细管法测定熔点实验关键是:
1、熔点管本身要干净,管壁不能太厚,封口要均匀。
2、毛细管装入样品要均匀,紧密。
3、装置要准确,达到一定位置。
4、 由于毛细管法是间接测熔点方法,所以加热升温速度是本实验的关键,应密切观察加热和熔化情况,当接近熔点10℃时升温速度一定要慢,应小于1~2℃/min;,注意观察温度变化。及时记下温度变化。
5、每个样品至少填装两支毛细管,平行测定两次。对于未知物可以先粗测一次,再精测二次。两次测定结果误差不大于±10C
6、测定下一个样品时让导热液慢慢冷却到样品近似熔点30℃以下左右,并换新的装有样品的毛细熔点管。
7.使用熔点测定管装置测定熔点时,应注意哪些事项?
(1)测定管口塞上的塞子应有缺口,使熔点测定管内与大气相通,避免测定管内的热浴液和空气由于受热时膨胀而冲开塞子或将测定管炸裂。
(2)插在塞子上的温度计的刻度应面向塞子缺口,便于观察温度
(3)温度计的水银球位于测定管的上下二侧管中间位置,因在此处加热对流循环好,温度均匀。
(4)毛细管内的样品中心与温度计的水银球中心位于同一水平面,温度计所显示的温度与样品的温度最为接近。
(5)导热液应装至测定管上侧管上方约1cm处。以免受热后膨胀溢出引起危险。另外,液面过高易引起毛细管熔点飘移,偏离温度计,影响测定的准确性。
(6)在熔点测定管的下侧管的外缘底部处进行加热。可使热浴液在测定管内进行对流循环,加热对密度小的热浴液沿着较细的侧管向上移动。在管径较大的直管处进行热量交换,由于此处的热浴液较多,热浴液的温度较为均匀,就会减小实验误差,如果在其他部位加热,受热的热浴液会向两侧传递热量,直管处温度变化过于强烈,不利于样品测定。
(五)数据记录和处理

二、有机化合物沸点的测定:
(一)沸点测定原理
什么叫沸点——当液体的蒸气压增大到与外界施于液面的总压力(通常是大气压力)相等时,就有大量气泡从液体内部逸出,即液体沸腾。这时的温度称为液体的沸点。
通常所说的沸点是指在101.3kPa下液体沸腾时的温度。在一定外压下,纯液体有机化合物都有一定的沸点,而且沸点变化在1-3℃,若含有杂质,则溶剂的蒸气压降低,沸点随之下降,,沸程也扩大,沸点变化范围(沸程)将超过3-5℃,所以测定沸点是鉴定有机化合物和判断物质纯度的依据之一,是衡量物质纯度的一个标准。但是具有固定沸点的液态有机物不一定都是纯的有机物,因为某些有机物常与其它组分形成二元或三元的共沸混合物,它们也有一定的沸点。测定沸点常用的方法有常量法(蒸馏法)和微量法(沸点管法)两种。
沸点测定装置图:见
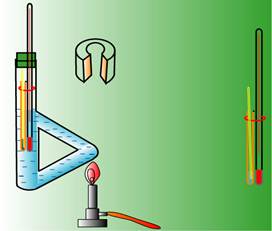
(二)实验步骤
微量法(沸点管法)沸点的测定:
方法一:取4~5滴待测样品滴入沸点管的外管中,将内管开口向下插入外管中,然后用小橡皮圈把沸点管附于温度计旁,小橡皮圈不能浸入导热液中,使装样品的中心位于温度计水银球的中部,再把该温度计的水银球位于b形管上下两叉管中间,然后加热。加热时由于气体膨胀,内管中会有小气泡缓缓逸出,当温度升到比沸点稍高时,管内会有一连串的小气泡快速而连续逸出,表明毛细管内压力超过了大气压。这时停止加热,使溶液自行冷却,气泡逸出的速度即渐渐减慢。在最后一气泡不再冒出并要缩回内管的瞬间记录温度,或者液体刚要进入毛细管、毛细管外液面与毛细管内液面等高时,即为毛细管内蒸气压与外界压力正好相等,此瞬间的温度即为该液体的沸点。待温度下降15~20℃后,可重新加热再测二次(3次所得温度数值不得相差±10C)
方法二:取4~5滴待测样品滴入沸点管的外管中,将内管开口向下插入外管中,然后用小橡皮圈把沸点管附于温度计旁,小橡皮圈不能浸入导热液中,使装样品的中心位于温度计水银球的中部,一起浸入小烧杯内水浴中加热,并用搅拌器不断上下搅动,使水浴温度均匀。加热时由于气体膨胀,内管中会有小气泡缓缓逸出,当温度升到比沸点稍高时,管内会有一连串的小气泡快速而连续逸出,表明毛细管内压力超过了大气压。这时停止加热,使溶液自行冷却,气泡逸出的速度即渐渐减慢。在最后一气泡不再冒出并要缩回内管的瞬间记录温度,或者液体刚要进入毛细管、毛细管外液面与毛细管内液面等高时,即为毛细管内蒸气压与外界压力正好相等,此瞬间的温度即为该液体的沸点。待温度下降15~20℃后,可重新加热再测二次(3次所得温度数值不得相差±10C)
按上述方法进行C2H5OH沸点测定并作出记录写出实验报告。
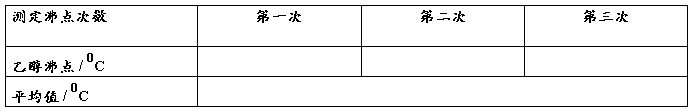
(三)数据记录和处理
沸点测定记录:
(四)注意事项:
1、 加热不能太快,待测液体不宜太少,以防样品全部汽化。
2、 内管里的空气要尽量赶尽。
3、 观察要仔细及时。
4、重新测定时应待导热液降温15~200C再测定下一次样品的沸点。各次测定误差不超过±10C
(五)正规写出实验报告—实验目的、原理、方法步骤、有关数据填写、分析与讨论,并请画出熔点沸点测定装置图,并对每一步装置加以说明。
习题:书上P144 1、2、3、4、5、
(第3题要写明熔点或沸点及熔程的影响)