药物分析实验指导
重庆医科大学药学系
药物分析教研室
二○##年六月
前 言
药物分析是一门综合性的应用学科。药物分析实验是药物分析课程的重要组成部分,是理论联系实际的重要环节。它的任务是培养学生树立药品质量观念,熟悉药品检验程序并培养学生具有检验常用的药物及制剂的能力。通过实验课的学习,学生应达到以下要求:
1.全面了解药物分析工作的程序及各环节的要求。
2 .掌握药物分析常用方法的原理及操作技术。
3.能运用本课程基本理论及有关专业知识分析和解决实验中的问题。
4.培养实事求是的科学态度和严谨认真的工作作风。
为此,要求学生实验前必须预习,明确实验目的,了解实验内容与方法,考虑实验中应注意的事项及安排实验的进程。实验中应认真操作,仔细观察实验现象并加以分析,作好原始记录,认真分析实验结果,力求做出准确可靠的结论。应遵守课堂纪律和实验室规则,注意安全、保持整洁。
本实验指导内容是根据药学专业教学计划要求,结合我校实际情况编写的。以药物分析中常用分析方法和典型药物及其制剂为主。使用时可根据学时数选择实验内容,也可作为教材的实例用作课堂教学的参考。
重庆医科大学药学系
药物分析教研室
二O##年六月
目 录
实验一 药物的杂质检查
(一) 标准溶液的配制
(二) 氯化钠的杂质检查
(三) 蒸馏水的杂质检查
实验二 苯甲酸钠的含量测定
实验三 阿司匹林片的分析
实验四 硫酸阿托品片的含量测定
实验五 复方磺胺甲噁 唑片的含量测定
实验六 盐酸普鲁卡因注射液的分析
实验七 维生素B1片的分析
实验八 维生素C注射液的分析
实验九 诺氟沙星含氟量的测定
实验十 诺氟沙星胶囊含量测定
实验十一 药房制剂的快速检验
实验十二 常用安眠镇静类药物的鉴别
实验十三 气色谱法测定酊剂中乙醇含量
实验十四 血浆中苯妥英浓度的高效液相色谱法测定
实验十五 高效液相色谱法测定阿莫西林胶囊的含量
实验十六 枸橼酸铁铵的杂质检查及含量测定
附录1~3 药品检验操作标准 原料药检验报告书
成品检验报告书
标准液配制及标定记录
实验一 药物的杂质检查
一、目的
1.了解药物杂质检查的意义。
2.掌握杂质检查的原理和方法。
3.掌握杂质限量的计算方法。
二、实验内容
(一)标准溶液的配制
1.标准氯化钠溶液的制备
称取氯化钠0.165g,置1000ml量瓶中,加水适量使溶解并稀释至刻度,摇匀,作为贮备液。临用前,精密量取贮备液10ml置100ml瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10μg的Cl)。
2.标准硫酸钾溶液的制备
称取硫酸钾0.18lg,置1000ml量瓶中,加水适量使溶解并稀释至刻度,摇匀,即得(每1ml相当于100μg的SO4)。
3.标准铁溶液的制备
称取硫酸铁铵 [FeNH4(SO4)2·12H2O] 0.863g,置1000ml量瓶中,加水溶解后,加硫酸2.5ml,用水稀释至刻度,摇匀,作为贮备液。
临用前,精密量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10μg的Fe)。
4.标准铅溶液的制备
称取硝酸铅0.160 g,置于1000ml量瓶中,加硝酸5ml与水50ml溶解后,用水稀释至刻度,摇匀,作为贮备液。
临用前精蜜量取贮备液10ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml相当于10μg的Pb)。
配制与贮存用的玻璃容器均不得含有铅。
5.标准砷溶液的制备
称取三氧化二砷0.132g,置1000ml量瓶中,加20%氢氧化钠溶液5ml溶解后,用适量的稀硫酸中和,再加稀硫酸10ml,用水稀释至刻度,摇匀,作为贮备液。
临用前,精密量取贮备液1ml,置100ml量瓶中,加稀硫酸1ml,用水稀释至刻度,摇匀,即得(每1ml相当于1.0μg的As)。
(二)氯化钠的杂质检查
1.酸碱度
取本品5.0g,加水50ml溶解后,加溴麝香草酚蓝指示液2滴。如显黄色,加氢氧化钠液(0.02mol/L)0.10ml,应变为蓝色;如显蓝色或绿色,加盐酸液(0.02mol/L)0.2ml,应变为黄色。
2.溶液的澄清度
取本品5.0g,加水25ml溶解后,溶液应澄清。
3.碘化物
取本品的细粉5.0g,置瓷蒸发皿内,滴加新配制的淀粉混合液 [取可溶性淀粉0.25g,加水2ml,搅匀,再加沸水至25ml,随加随搅拌,放冷,加硫酸液(0.025mol/L)2ml,亚硝酸钠试液3滴与水25ml,混匀] 适量使晶粉湿润,置日光下(或日光灯下)观察,5分钟内晶粒不得显蓝色痕迹。
4.溴化物
取本品2.0g,加水10ml使溶解,加盐酸3滴与氯仿1ml,边振摇边滴加2%氯胺T溶液(临用新制)3滴,氯仿层如显色,与标准溴化钾溶液(精密称取在105℃干燥至恒重的溴化钾0.1485g,加水使溶解成100ml,摇匀)1.0ml用同一方法制成的对照液比较,不得更深。
5.硫酸盐
取本品5.0g,加水溶解成约40ml(溶液如显碱性,可滴加盐酸使成中性),溶液如不澄清,应滤过,置50ml纳氏比色管中,加稀盐酸2ml,摇匀,即得供试溶液。另取标准硫酸钾溶液1.0ml置50ml纳氏比色管中,加水使成约40ml,加稀盐酸2ml摇匀,即得对照溶液。于供试溶液与对照溶溶中,分别加入25%氯化钡溶液5ml,用水稀释使成50ml,充分摇匀,放置10分钟,同置黑色背景上,从比色管上方向下观察、比较、供试品管浊度不得更浓。
6.钡盐
取本品4.0g,加水20ml,溶解后,滤过,滤液分为两等份,1份中加稀硫酸2ml,另一份中加水2ml,静置15分钟,两液应同样澄清。
7.钙盐
取本品2.0g,加水10ml使溶解,加氨试液1ml,摇匀,加草酸铵试液1ml,5分钟内不得发生浑浊。
8.镁盐
取本品1.0g,加水20ml使溶解,加氢氧化钠试液2.5ml与0.05%太坦黄溶液0.5ml,摇匀,产生的颜色与标准镁溶液(精密称取在800℃炽灼至恒重的氧化镁16.58mg,加盐酸2.5ml与水适量使溶解成1000ml,摇匀)1.0ml,用同一方法制成的对照液比较,不得更深。
9.钾盐
取本品5.0g,加水20ml溶解后,加稀醋酸2滴,加四苯硼钠溶液(取四苯硼钠1.5g,置乳钵中,加水10ml研磨后,再加水40ml,研匀,用质密的滤纸滤过,即得)2ml,加水使成50ml,如湿浑浊,与标准硫酸钾溶液12.3ml用同一方法制成的对照液比较,不得更浓。
10.干燥失重
取供试品,混合均匀(如为较大的结晶,应先迅速捣碎,使成2mm以下的小粒)。分取约lg,平铺在130℃干燥至恒重的扁形称瓶中,厚度不超过5mm,精密称定。将瓶盖取下,置称瓶旁,或将瓶盖半开,置烘箱内于130℃干燥约2~3小时后,将称瓶盖好,取出,置干燥器中放置40分钟后,精密称定重量。再按上述方法自“将瓶盖取下……”起,继续干燥1~2小时,依法操作,至恒重为止。从减失重量和取样量计算供试品的干燥失重(规定不得超过0.5%)。
11.铁盐
取本品5.0g,加水溶解成25ml,置50ml纳氏比色管中,加稀盐酸4ml与过硫酸铵50mg,用水稀释成35ml后,加30%硫氰酸铵溶液3ml,再加水适量稀释成50ml,摇匀。如显色,立即与标准铁溶液1.5ml用同一方法制成的对照液比较,不得更深。
12.重金属
取本品5.0g,加水20ml溶解后,置25ml纳氏比色管中,加醋酸盐缓冲液(pH3.5)2ml与水适量使成25ml,加硫代乙酰胺试液(临用前取5.0ml混合液,加4%硫代乙酰胺溶液1ml,水浴加热20秒,冷却,立即使用)2ml,摇匀,放置2分钟,置白纸上,自上向下透视。如显色,立即与标准铅溶液1.0ml用同一方法的对照液比较,不得更深。
13.砷盐
仪器装置:如图1,A为100ml标准磨口锥形瓶,B为中空的标准磨口塞,上连导气管C(外径8.0mm,内径6.0mm),全长约180mm。D为具孔的有机玻璃旋塞,其上部为圆形平面,中央有一圆孔,孔径与导气管C的内径一致,其下部孔径与导气管C的外径相适应。将导气管C的顶端导入旋塞下部孔内,并使管壁与旋塞的圆孔恰相吻合,粘合固定,E为中央具有圆孔(孔径6.0mm)的有机玻璃旋塞盖,与D紧密吻合。
测试时,于导气管C中装入醋酸铅棉花60mg(装管高度约60~80mm),再于旋塞D的顶端平面上放一片溴化汞试纸(试纸大小以能覆盖孔径而不露出平面外为宜),盖上旋塞E并旋紧,即得。
标准砷斑的制备:精密量取标准砷溶液2ml,置A瓶中,加盐酸5ml与水21ml,再加碘化钾试液5ml与酸性氯化亚锡试液5滴,在室温放置10分钟后,加锌粒2g,立即照上法装妥的导气管C密塞于A瓶上,并将A瓶置25~40℃水浴中,反应45分钟后,取出溴化汞试纸,即得。
检查法,取本品5.0g,置A瓶中,加水23ml溶解后,加盐酸5ml,照标准砷斑的制备,自“加碘化钾试液5ml”起,依法操作,将生成的砷斑与标准砷斑比较,不得更深。

(三)蒸馏水的杂质检查
1.酸碱度
取本品10ml,加甲基红指示液2滴,不得显红色;另取10ml,加溴麝香草酚蓝指示液5滴,不得显蓝色。
2.氯化物、硫酸盐与钙盐
取本品,分置3支试管中,每管各50ml。第一管中,加硝酸5滴与硝酸银试液1ml;第二管中,加氯化钡试液2ml,第三管中,加草酸铵试液2ml,均不得发生浑浊。
3.硝酸盐与亚硝酸盐
取本品15ml,加醋酸液(6mol/L)18ml,对氨基苯磺酸-α-萘胺试液2ml,锌粉0.01g,摇匀,放置15分钟,不得显粉红色。
4.氨
取本品50ml,加碱性碘化汞钾试液2ml,放置15分钟;如显色,与氯化铵溶液(取氯化铵31.5mg,加无氨蒸馏水适量使溶解并稀释成1000ml)2.0ml,加无氨蒸馏水48ml与碱性碘化汞钾试液2ml制成的对照液比较,不得更深。
5.二氧化碳
取本品25ml,置50ml具塞量筒中,加氢氧化钙试液25ml,密塞振摇,放置,1小时内不得发生浑浊。
6.易氧化物
取本品100ml,加稀硫酸10ml,煮沸后,加高锰酸钾溶液(0.02mol/L)0.1ml,再煮沸10分钟,粉红色不得完全消失。
7.不挥发物
取本品100ml,置105℃恒重的蒸发皿中,在水浴上蒸干,并在105℃干燥至恒重,遗留残渣不得超过1mg。
8.重金属
取本品40ml,加醋酸盐缓冲液(pH3.5)2ml,硫代乙酰胺试液(临用前取5.0ml混合液,加4%硫代乙酰胺溶液1ml,水浴加热20秒,冷却,立即使用。)2ml,摇匀,放置2分钟,与本品42ml与醋酸盐缓冲液(pH3.5)2ml的混合液比较,颜色不得更深。
三、说明
1.药物杂质检查必须注意按规定的检查条件,严格遵守平行原则。平行原则是指样品与标准必须在同一条件下进行反应与比较。即应尽量选择容积、口径和色泽相同的比色管,在同一光源、同衬底上,以相同的方式(一般是自上而下)观察,所加入的试药种类、用量、加入顺序和反应时间等也应一致。
2.杂质限量是指药物中允许存在杂质的最大量。其定义式为:
杂质限量= 
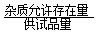 ×100%
×100%
3.药物的杂质检查是限度检查,合格者仅说明其杂质量在药品质量标准允许范围内,并不说明药品中不含该项杂质。
四、思考题
1.杂质检查的意义是什么?
2.药物中杂质来源主要有哪些途径? 什么是一般杂质? 什么是特殊杂质?
3.药物杂质检查应严格遵循什么原则?
4.试计算出氯化钠中溴化物、硫酸盐、镁盐、钾盐、重金属和砷盐的限量。
5.某一药物取样0.5g进行重金属检查,药典规定限量不得超过10ppm,应取多少毫升标准铅溶液?
6.某一药物规定砷盐限量不得超过4ppm,取标准砷溶液2ml对照,问应取供试品多少克?
实验二 苯甲酸钠的含量测定
一、目的
掌握双相滴定法测定苯甲酸钠含量的原理和操作
二、操作
取本品1.5g,精密称定,置分液漏斗中,加水约25ml,乙醚50ml与甲基橙指示液2滴,用盐酸滴定液 (0.5mol/L)滴定,随滴随振摇,至水层显持续橙红色,分取水层,置具塞锥形瓶中,乙醚层用水5ml洗涤,洗涤液并入锥形瓶中,加乙醚20ml,继续用盐酸滴定液(0.5mol/L)滴定,随滴随振摇,至水层显持续橙红色,即得,每1ml的盐酸滴定液(0.5mol/L)相当于72.06mg的C7H5O2Na。
本品按干燥品计算,含C7H5O2Na不得少于99.0%
三、说明
1.苯甲酸钠为有机酸的碱金属盐,显碱性,可用盐酸标准液滴定。
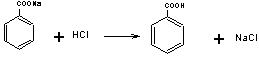
在水溶液中滴定时,由于碱性较弱(Pkb=9.80)突跃不明显,故加入与水不相溶混的溶剂乙醚提除反应生成物苯甲酸,使反应定量完成,同时也避免了苯甲酸在瓶中析出影响终点的观察。
2.滴定时应充分振摇,使生成的苯甲酸转入乙醚层。
3.在振摇和分取水层时,应避免样品的损失,滴定前,应用乙醚检查分液漏斗是否严密。
四、思考题
1.乙醚为什么要分两次加入?第一次滴定至水层显持续橙红色时,是否已达终点?为什么?
2.分取水层后乙醚层用5ml水洗涤的目的是什么?
实验三 阿司匹林片的分析
一、目的
1.掌握片剂分析的特点及赋形剂的干扰与排除方法。
2.掌握阿司匹林片鉴别、检查、含量测定的原理及方法。
二、操作
[鉴别]
1.取本品的细粉适量(约相当于阿司匹林0.1g),加水10ml煮沸,放冷,加三氯化铁试液1滴,即显紫堇色。
2.取本品的细粉(约相当于阿司匹林0.5g),加碳酸钠试液10ml,振摇后,放置5分钟,滤过,滤液煮沸2分钟,放冷,加过量的稀硫酸,即析出白色沉淀,并发生醋酸的臭气。
[检查]
游离水杨酸
取本品的细粉适量(约相当于阿司匹林0.1g),加无水氯仿3ml,不断搅拌2分钟,用无水氯仿湿润的滤纸滤过,滤渣用无水氯仿洗涤2次,每次1ml,合并滤液与洗液,在室温下通风挥发至干;残渣用无水乙醇4ml溶解后,移至100ml量瓶中,用少量5%乙醇洗涤容器、洗液并入量瓶中,加5%乙醇稀释至刻度,摇匀,分取50ml,立即加新制的稀硫酸铁铵溶液 [取盐酸液(1mol/L)1ml,加硫酸铁铵指示液2ml后,再加水适量使成100ml] 1ml,摇匀;30秒钟内如显色,与对照液(精密称取水杨酸0.1g,置1000ml量瓶中,加冰醋酸1ml,摇匀,再加水适量至刻度,摇匀,精密量取1.5ml,加无水乙醇2ml与5%乙醇使成50ml,再加上述新制的稀硫酸铁铵溶液1ml,摇匀)比较,不得更深。
溶出度
取本品1片,照溶出度测定法第一法(20##年药典二部附录),以稀盐酸24ml加水至1000ml为溶剂,转蓝转速为每分钟100±5转,依法操作,经30分钟时,取溶液10ml滤过,精密量取续滤液3ml置50ml量瓶中,加0.4%氢氧化钠液5ml,置水浴中煮沸5分钟,放冷,加硫酸液2.5ml;并加水稀释至刻度,摇匀。照分光光度法,在303nm的波长处测定吸收度,按C7H6O3的吸收系数E1%1cm)为265计算,再乘以1.304,计算出每片的溶出量;不得少于标示量的80%。其他应符合片剂项下有关的各项规定(20##年中国药典二部)。
[含量测定]
取本品10片,精密称定,研细,精密称出适量(约相当于阿司匹林0.3g),置锥形瓶中,加中性乙醇(对酚酞指示液显中性)20ml,振摇,使阿司匹林溶解,加酚酞指示液3滴,滴加氢氧化钠滴定液(0.1mol/L)至溶液显粉红色,再精密加氢氧化钠滴定液(0.1mol/L)40ml,置水浴上加热15分钟并时时振摇,迅速放冷至室温,有硫酸滴定液(0.05mol/L)滴定,并将滴定的结果用空白试验校正。每1ml氢氧化钠滴定液(0.1mol/L)相当于18.02mg的C9H8O4。
本品含阿司匹林(C9H8O4)应为标示量的95.0—105.0%。
三、说明
1.杨酸与三氯化铁试液显紫堇色。

2.检查溶出度是利用在碱性条件下,加热,使阿司匹林水解,测定水解产物水杨酸的吸收度,按E1%1cm为265计算水杨酸的含量,故用阿司匹林的含量表示溶出度时应乘以换算因数。
换算因数=MASP/MSA=1.304
3.为消除阿司匹林水解产物水杨酸,醋酸及稳定剂枸椽酸、酒石酸对含量测定的影响故采用两步滴定法。
第一次滴定:CH3COOH+NaOH→CH3COONa+H2O

第二次滴定:

四、思考题
1.简述鉴别试验的原理。
2.检查游离的水杨酸时,为防止阿司匹林水解,操作中应注意哪些问题? 计算供试品中阿司匹林的限量。
3.含量测定时,为什么要作空白试验?空白试验的操作如何进行? 为什么加中性乙醇溶解供试品?如何制备中性乙醇?
4.你认为含量测定时,应注意哪些问题才可以获得准确的结果?
实验四 硫酸阿托品片的含量测定
一、目的
1.掌握酸性染料比色法的基本原理和操作。
2.掌握比色法的基本方法,要求和计算。
二、实验内容
1.对照品溶液的制备
精密称取在120℃干燥至恒重的硫酸阿托品对照品25mg,置25ml量瓶中,加水溶解并稀释至刻度,摇匀;精密量取5ml,置100ml量瓶中,加水稀释至刻度,摇匀,即得(每1ml含无水硫酸阿托品50μg)。
2.供试品溶液的制备
取本品20片,精密称定、研细,精密称出适量(约相当于硫酸阿托品2.5mg),置50ml量瓶中,加水振摇使硫酸阿托品溶解并稀释至刻度,用干燥滤纸滤过,弃去初滤液,收集续滤液,即得。
3.测定法
精密量取对照品溶液与供试品溶液各2ml,分别置于预先精密加入氯仿10ml的分液漏斗中,各加溴甲酚绿溶液 [取溴甲酚氯50mg与邻苯二甲酸氢钾1.021g,加氢氧化钠液(0.2mol/L)6.0ml使溶解,再加水稀释至100ml,摇匀,必要时滤过] 2.0ml,振摇提取2分钟后,静置使分层,分取澄清的氯仿液,置1cm吸收池中,在420nm的滤长处分别测定吸收度,计算,并将结果与1.027相乘,即得供试量中硫酸阿托品(C17H23NO3)2·H2SO4·H2O]的重量。
本品含硫酸阿托品[(C17H23NO3)2·H2SO4·H2O]应为标示量的90.0~110.0%。
三、说明
1. 硫酸阿托品的结构:

2.本实验采用酸性染料比色法测定硫酸阿托品含量,实验中应严格控制水相pH并保证离子对化合物能定量提取进入氯仿层。
3.分液漏斗活塞处宜涂甘油淀粉作润滑剂,其配制方法是:取甘油22g,加入可溶性淀粉9g,混匀,加热至140℃,保持30分钟,并不断搅拌至透明,放冷,即得。
4.振摇提取时既要能定量地将离子对化合物提入氯仿层,又要防止乳化和少量水份不混入氯仿层,因此,需小心充分振摇,并使静置分层后再分取氯仿层,同时可在分液漏斗颈部放置少许脱脂棉以吸附氯仿中少量水份。
四、思考题
1.试述酸性染料比色法的原理。
2.酸性染料比色法的主要条件有哪些? 结合实验说明如何控制这些条件。
3.应如何作空白试验?
4.校正因子1.027是怎样算得的?
实验五 复方磺胺甲噁唑片的含量测定
一、目的
1.掌握复方制剂的分析特点及赋形剂的干扰与排除方法。
2.掌握双波长分光光度法测定复方磺胺甲噁 唑片中磺胺甲噁唑与甲氧苄啶含量的原理与方法。
二、实验内容
1.供试品溶液的制备
取本品10片,精密称定,研细,精密称取适量(约相当于磺胺甲噁 唑50mg与氧苄啶10mg),置于100ml量瓶中,加乙醇适量,振摇15分钟磺胺甲口恶 唑与甲氧苄啶溶解,加乙醇稀释至刻度,摇匀,滤过,取续滤液作为供试品溶液。
2.对照品溶液的制备
精密称取105℃干燥至恒重的磺胺甲噁 唑对照品50mg与甲氧苄啶对照品10mg,分置100ml量瓶中,各加乙醇溶解并稀释至刻度,摇匀,分别作为对照品溶液(1)与对照品溶液(2)。
3.磺胺甲噁 唑的测定
精密量取供试品溶液与对照品溶液(1)、(2)各2ml,分置100ml量瓶中,各加0.4%氢氧化钠溶液稀释至刻度,摇匀。取对照品溶液(2)的稀释液;以257nm为测定波长(λ2), 在304nm波长附近(每间隔0.5nm)选择等吸收点波长为参与波长(λ1),要求△A=Aλ2-Aλ1=0。再在λ2与λ1波长处分别测定供试品溶液的稀释液与对照品溶液(1)的稀释液的吸收度,求出各自的吸收度差值(△A),计算,即得。
4.甲氧苄啶的测定
精密量取上述供试品溶液与对照品溶液(1)(2)各5ml,分置100ml量瓶中,各加盐酸一氯化钾溶液 [取盐酸液(0.1mol/L)75ml与氯化钾6.9g,加水至1000ml摇匀] 稀释至刻度,摇匀。取对照品溶液(1)的稀释液,以239.0nm为测定波长(λ2),在295nm波长附近(每间隔0.2nm)选择等吸收点波长为参比波长(λ1),要求△A=Aλ2-Aλ1=0。再在λ2与λ1波长处分别测定供试品溶液的稀释液与对照品溶液(2)的稀释液的吸收度,求出各自的吸收度差值(△A),计算,即得。
本品每片中含磺胺甲噁 唑(C10H11N3O2S)应为0.360~0.440g,含甲氧苄啶(C14H12N4O3)应为72.0~88.0mg。
三 说明
1.磺胺甲噁 唑与甲氧苄啶的结构分别为:
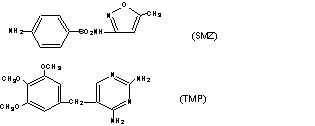
SMZ与TMP的紫外吸收图谱分别为:

图1.SMZ的紫外吸收图谱 图2.TMP的紫外吸收图谱
1 TMP(2.0μg/ml); 2 SMZ(10.0μg/ml) 1 TMP(5.0μg/ml); 2 SMZ(25.0μg/ml)
3 SMZ+TMP; 4 辅料 3 SMZ+TMP; 4 辅料
2.复方磺胺甲噁唑片的处方为:
磺胺甲噁唑 400g
甲氧苄啶 80g 制成1000片
3.本实验采用双波长分光光度法测定SMZ和TMP的含量。由于干扰组分在测定波长和参比波长处吸收度相等,可在计算△A时消去,所以应用本法可以不经分离,直接测定复方制剂中SMZ和TMP的含量。
四、思考题
1.试简述双波长分光光度法的原理。
2.试查阅本品的其它分析方法,从中了解复方制剂分析的特点及发展趋势。
实验六 盐酸普鲁卡因注射液的分析
一、目的
1.掌握用薄层层析法检查盐酸普鲁卡因注射液中对氨基苯甲酸限量的原理及方法。
2.掌握用重氮化滴定法、分光光度法测定盐酸普鲁卡因含量的原理及方法。
二、实验内容
(一)鉴别 取本品适量(约相当于盐酸普鲁卡因50mg),加稀盐酸1ml,加0.1mol/L亚硝酸钠溶液数滴,滴加碱性β-萘酚试液数滴,生成橙红色沉淀。
(二)对氨基苯甲酸的检查
薄层层析法:精密量取本品,加乙醇稀使成每1ml中含盐酸普鲁卡因2.5mg的溶液,作为供试品溶液,另取对氨基苯甲酸对照品,加乙醇制成每1ml含30μg的溶液,作为对照品溶液,吸取上述两种溶液各10μl,分别点于含有羧甲基纤维素钠为粘合剂的硅胶H薄层板上,用苯—冰醋酸—丙酮—甲醇(14∶1∶1∶4)为展开剂,展开后,取出晾干,用对一二甲氨基苯甲醛溶液〔2%对一二甲氨基苯甲醛乙醇溶液100ml,加入冰醋酸5ml制成〕喷雾显色。供试溶液如显与对照品相应的杂质斑点,与对照品溶液的主斑点比较,不得更深。
薄层板的制备:取硅胶H 2g,加0.5%羧甲基纤维素钠水溶液适量调成糊状,均匀涂布于玻璃板(5×15cm)上,置水平台上,在空气中晾干,再于110℃烘0.5—1小时,放置干燥器中备用。
(三)含量测定
1.重氮化法:调节永停滴定仪上电阻R使加于电极上电压约为50mV。精密量取本品适量(约相当于盐酸普鲁卡因0.1g),置烧杯中,加水40ml与盐酸液(1→2)15ml,加溴化钾2g,置电磁搅拌器上,插入铂一铂电极后,在15—20℃,将滴定管的尖端插入液面下约2/3处,用亚硝酸液(0.05mol/L)迅速滴定,随滴随搅拌,至近终点时,将滴定管的尖端提出液面,用少量水淋洗尖端,洗液并入溶液中,继续缓缓滴定,至电流计指针突然偏转,并不再回复,即为滴定终点。每1ml的亚硝酸钠液(0.05mol/L)相当于13.64mg的C13H20N2O2·HCl。
本品含盐酸普鲁卡因应为标示量的95.0—105.0%。
2.分光光度法:
精密量取本品1ml于100ml量瓶中,加水至刻度,摇匀,精密量取此稀释液10ml于另一100ml量瓶中,加水至刻度(即每1ml含盐酸普鲁卡因10μg),摇匀,照分光光度法在290nm的波长处测定吸收度,按C13H20N2O2·HCl的吸收系数(E1%1cm)为680计算,即得。
三、说明
1.盐酸普鲁卡因,对氨基苯甲酸在冰醋酸中与对二甲氨基苯甲醛缩合而呈色。

四、思考题
1. 计算用TLC法检查对氨基苯甲酸的限量。
实验七 维生素B1片的分析
一、目的
1.掌握维B1鉴别反应的原理和方法;
2. 掌握紫外分光光度法测定药物含量的原理和方法。
二、实验内容
(一) 鉴别
取本品的细粉适量,加水搅拌,滤过,滤液蒸干。
(1)取残渣约5mg,加氢氧化钠试液2.5ml溶解后,加铁氰化钾试液0.5ml与正丁醇5ml,强力振摇2分钟,放置使分层,上面的醇层显强烈的蓝色荧光;加酸使成酸性,荧光即消失;再加碱使成碱性,荧光又显出。
(2)取残渣少许,加水溶解,加硝酸使成酸性后,加硝酸银试液,即生成白色凝乳状沉淀;分离,沉淀加氨试液即溶解,再加硝酸,沉淀复生成。
(3)取残渣少许,置试管中、加等量的二氧化锰,混匀,加硫酸湿润,缓缓加热,即发生氯气,能使湿润的碘化钾淀粉试纸显蓝色。
(二)含量测定
取本品20片,精密称定,研细,精密称取适量(约相当于维生素B1 25mg),置100ml量瓶中,加盐酸溶液(9→1000ml)约70ml,振摇15分钟使维生素B1溶解,加盐酸溶液(9→1000)稀释至刻度,摇匀,用干燥滤纸滤过,精密量取续滤液5ml,置另一100ml量瓶中,再加盐酸溶液(9→1000)稀释至刻度,摇匀,照分光光度法,在246nm的波长处测定吸收度,按C12H17ClN4OS·HCl的吸收系数(E1%1cm)为421计算,即得。
本品含维生素B1(C12H17ClN4OS·HCl)应为标示量的90.0~110.0%。
三、思考题
1.试简述以上维生素B1鉴别反应的原理。
2.试简述吸收系数测定药物含量的特点和一般方法。
3.如何计算供试品中维生素B1的含量。
实验八 维生素C注射液的分析
一、目的
1.掌握注射液分析的特点及附加成分的干扰与排除方法。
2.掌握维生素C注射液鉴别和含量测定的原理和方法。
二、实验内容
(一)鉴别
取本品适量(约相当于维生素C 0.2g),加水稀释至10ml,照下述方法试验。
1.取溶液5ml,加硝酸银试液0.5ml,即生成银的黑色沉淀。
2.取溶液5ml,加二氯靛酚钠度液1—2滴,试液的颜色即消失。
(二)含量测定
1.碘量法
精密量取本品适量(约相当于维生素C 0.2g),加水15ml与丙酮2ml,摇匀,放置5分钟,加稀醋酸4ml与淀粉指示液1ml,用碘液(0.1mol/L)滴定,至溶液显蓝色并持续30秒钟不褪,即得。每1ml的碘液(0.1mol/L)相当于8.806mg的C6H8O6。
本品含维生素C应为标示量的90.0~110.0%。
2.比色法
(1)标准曲线的制备:
精密称取维生素C对照品约100mg,置100ml量瓶中,加水溶解并稀释至刻度,摇匀。精密量取上述溶液5ml置另一100ml量瓶中,加水稀释至刻度,摇匀,即得(50μg/ml)。
分别精密量取以上对照品溶液2、3、4、5、6ml,分置25ml量瓶中,加水稀释至10ml,加1,10—菲咯啉—铁(Ⅲ)试液[取1,10—菲咯啉(一水物)0.198g,加盐酸(1mol/L)2ml和硫酸铁铵0.16g,加水溶解并稀释至100ml。]1ml,混匀,加水稀释至刻度,摇匀,于510nm波长处测定吸收度。用吸收度对对照品量(mg)回归,即得。
(2)维生素C注射液的含量测定
精密量取本品适量(约相当于维生素C 500mg),置100ml量瓶中,加水稀释至刻度,摇匀。精密量取2ml置另一100ml量瓶中,加水稀释至刻度,摇匀,即得(100μg/ml)。
精密量取供试品溶液2ml于25ml量瓶中,加水稀释至10ml,加入1,10—菲咯啉—铁(Ⅲ)试液1ml,以下按照标准曲线方法操作。根据供试品溶液的吸收度和标准曲线计算出维生素C的含量,即得。
三、说明:
1. 在酸性溶液中铁(Ⅲ)氧化抗坏血酸生成去氢抗坏血酸,同时铁(Ⅲ)被还原成亚铁离子,后者与1,10—菲咯啉络合生成红色的亚铁菲咯啉离子〔Fe(C12H8N2)3〕2+,在波长510mm处有最大吸收。可用作抗坏血酸及其制剂的含量测定。反应式为:

2.经试验,注射剂中抗氧剂焦亚硫酸钠、亚硫酸钠等不干扰测定。
四、思考题
1.试简述维生素C的结构与分析方法之间的关系。
2.用碘量法测定含量时,加入丙酮和稀醋酸的目的是什么?
3.用比色法测定维生素C含量时,空白对照溶液如何制备? 实验中应注意哪些问题,才能保证实验结果的准确?
实验九 诺氟沙星(氟哌酸)含氟量的测定
一、目的
1.掌握氧瓶燃烧法的操作方法。
2.掌握茜素氟蓝比色法的原理和方法。
二、实验内容
1.仪器装置
燃烧瓶为500ml磨口、硬质玻璃锥形瓶(或根据取样量选用250mol、100ml或2000ml的磨口、硬质玻璃锥形瓶)瓶塞应严密,空心,底部熔封铂丝一根(直径为1mm),铂丝下端作成网状或螺旋状,长度约为瓶身长度的2/3,如图A。
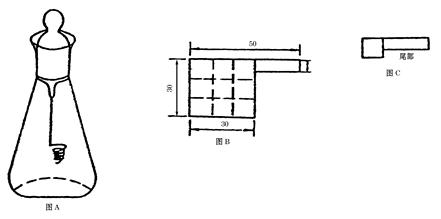
2.供试品的燃烧破坏及供试溶液的配制
取本品细粉35mg,精密称定。置于无灰滤纸(如图B)中心,按虚线折叠(如图C)后,固定于铂丝下端的网内或螺旋处,使尾部露出。另在燃烧瓶内加水20ml,作为吸收液,并将瓶颈用水湿润,小心急速通入氧气约1分钟(通气管应接近液面,使瓶内空气排尽),立即用表面皿覆盖瓶口,移至他处;点燃包有供试品的滤纸尾部,迅速放入燃烧瓶中,按紧瓶塞,俟燃烧完毕(应无黑色碎片),用少量水封闭瓶口,充分振摇,使生成的烟雾完全吸入吸收液中,放置15分钟,用水冲洗瓶塞及铂丝,合并洗液及吸收液,转移至100ml量瓶中,用水洗净燃烧瓶,洗液并入量瓶中,加水稀释至刻度,摇匀,作为供试溶液。
3.氟对照溶液的制备
精密称取经105℃干燥1小时的氟化钠22.1mg,置100ml量瓶中,加水溶解并稀释至刻度,摇匀;临用前,精密量取10ml,置另一50ml量瓶中,加水稀释刻度,摇匀,即得。(每1ml相当于20μg的F)。
4.比色测定
精密量取供试溶液与对照溶液各2ml,分别置50ml量瓶中,各加茜素氟蓝试液10ml,摇匀,再各加12%醋酸钠的稀醋酸溶液3.0ml与硝酸亚铈试液10ml,加水稀释至刻度,摇匀,在暗处放置1小时,置1cm吸收池中,在610nm的波长处分别测定吸收度,并将供试液的吸收度用空白试验校正,计算,即得。含氟量应不少于5.6%。
三、说明
1.氧瓶燃烧法系将有机药物在充满氧气的燃烧瓶中燃烧破坏,使有机结合的测定元素转化为无机的离子状态,俟燃烧产物被吸收液完全吸收后,再采用适当的方法来鉴别、检查或测定卤素、硫、硒、磷等元素的方法。
2.本实验的原理是将诺氟沙星用氧瓶燃烧法燃烧破坏,使有机结合的氟转变为无机的F-,再用茜素氟蓝比色法测定其含量。
3.本实验的关键在于供试品是否燃烧完全和吸收完全。
4.本实验应有防护措施,如戴防护罩、眼镜等。
5.比色法中的空白试验,是取同体积的溶剂代替供试品溶液或对照品溶液,然后在与供试品溶液和对照品溶液完全平行的条件下加入有关试剂,作为空白对照溶液(又称试剂空白)。测定吸收度时,先将空白对照溶液的吸收度作为零(或透光率为100%),再测定供试品溶液和对照品溶液的吸收度。
四、思考题
1.为使供试品燃烧完全和吸收完全,应注意哪些问题?
2.氧瓶燃烧法有哪些优点? 有什么不足?
3.写出供试品中氟含量的计算公式。已知诺氟沙星(C16H18FN3O3)的分子量为319.24,氟(F)的原子量为19.00,试计算诺氟沙星的理论含氟量。
实验十 诺氟沙星(氟哌酸)胶囊含量测定
一、目的
1.掌握非水滴定法的原理及操作;
2.熟悉非水滴定在药物分析中的应用。
二、操作
1.诺氟沙星的鉴别
(1)取本品内容物适量(约相当于诺氟沙星0.15g)置干燥试管中,加丙二酸0.1g与醋酐2ml,振摇,并在80-90℃水浴中加热10-15mm,显红棕色。
(2)取本品内容物适量,加0.4%氢氧化物溶液适量,振摇使诺氧沙星溶解,稀释成每1ml中含诺氟沙星5μg,滤过,充去初滤液,取续滤液,在273,325与336nm波长处测定有最大吸收。
2.含量测定 精密称取本品内容物适量(约相当于诺氟沙星0.25g),加冰醋酸30ml,振摇使诺氟沙星溶解,加橙黄Ⅳ指示液10滴,用高氯酸液(0.1mol/L)滴定,至溶液显紫红色,并将滴定的结果用空白试验校正。
三、说明
1.ChP1995版规定本品含诺氟沙星应为标示量的90.0—110.0%,滴定时,每1ml0.1mol/L的高氯酸液相当于31.93mg的C16H18FN3O3。
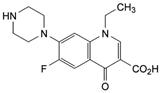

2. 0.1mol/L的高氯酸溶液的配制及标定参见ChP1995版附录。高氯酸不稳定,注意避光保存。
3.水的体膨胀系数较小(0.21×10-3/C0),一般酸碱标准溶液的浓度受室温改变的影响不大。而多数有机溶剂的体膨胀系数较大,例如,冰醋酸的体膨胀系数为(1.1×10-3/C0)其体积随温度改变较大。所以高氯酸冰醋酸溶液滴定样品时和标定时温度若有差别,则应重新标定或按下式将标准溶液的浓度加以校正:
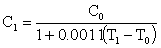
式中:0.0011为冰醋酸的体膨胀系数,t0为标定时的温度,t1为测定时的温度,C0为标定时的浓度,C1为测定时的浓度。
4.现行药典对本品含量测定的方法为HPLC法。
四、思考题
非水滴定应用于药物分析有何特点? 哪几类药物适合用非水滴定法测定其含量。
实验十一 药房制剂的快速检验
一、目的
1.了解快速检验的特点。
2.熟悉容量分析,比色分析在医院药房制剂检验中的应用。
二、实验内容
(一)氯化钠注射液含量的快速检验
精密量取本品10ml,加水40ml,加糊精溶液(1→50)5ml与荧光黄指示液5-8滴,用硝酸银液(0.1mol/L)滴定,随滴随振摇,至14.54ml时,应无明显变色,至16.26ml时,应为微红色,(每1ml0.1mol/L AgNO3溶液相当于5.844mg的NaCl)即合格品应耗0.1mol/L AgNO3溶液的限度为14.54-16.26ml。(按药典规定本品含氯化钠应为0.850~0.950%)
(二)3%硼酸溶液的快速检验
精密量取本品5ml,置25ml量瓶中,加水稀释至刻度,摇匀,精密量取2ml,加中性甘油5ml,水4ml,酚酞指示液3滴,用氢氧化钠滴定液(0.1mol/L)滴定,至××ml时,应无明显淡红色;至××ml时,应呈明显淡红色。
(三)5%葡萄糖注射液的快速检验
1.旋光法:
精密量取本品适量(约相当于葡萄糖10g),置100ml量瓶中,加氨试液0.2ml(10%或10%以下规格的本品可直接取样测定),用水稀释至刻度,摇匀,静置10分钟,测定旋光度与2.0852相乘,即得供试品量中含有C6H12O6·H2O的重量(g)。
2.碘量法:
精密量取本品2ml,置25ml量瓶中,加水稀释至刻度,摇匀。吸取5.00ml置碘量瓶中,准确加入碘滴定液(0.1mol/L)5ml,再分二次加入氢氧化钠滴定液(0.1mol/L),每次5ml,密塞,振摇约10秒种,加完后,密塞,于暗处放置10分钟,加稀硫酸1ml,用硫代硫酸钠滴定液(0.1mol/L)滴定,至2.88ml时,浅黄色不应消失,至3.09ml时,应为无色溶液。
三、说明
1.为了保证医院药房自配制剂的质量,常采用溶量分析的方法测定制剂中各组分含量,其方法为:根据各种制剂的处方及上下限要求(一般为±5%),按取样量预先计算出应该消耗标准溶液毫升数的范围,实验时,实际用量在此范围内含量为合格,不必再行计算。
消耗标准溶液毫升数(V)的计算:
V=G/T 下限=V×95%
上限=V×105%
G:供试品中应含被测组分的量,即标示量。
T:每1ml标准溶液相当于被测组分的重量,即滴定度。
2.硼酸含量测定原理:
C3H5(OH)3+H3BO3→C3H5(OH)HBO3+2H2O
Ka=7.3×10 -10
Ka=3×10 -7
C3H5(OH)HBO3+NaOH→C3H5(OH)NaBO3+H2O
3.葡萄糖含量测定原理
I2+2NaOH→NaIO+NaI+H2O
CH2OH(CHOH)4CHO+NaIO+NaOH→CH2OH(CHOH)4COONa+NaI+H2O
3NaIO(剩余) NaIO3+2NaI
NaIO3+2NaI
NaIO3+6HCl+5NaI→3I2+6NaCl+3H2O
I2+2Na2S2O3→2NaI+Na2S4O6
计算公式:C=100α / [α]d 按药典规定, 葡萄糖的[α]tD为52.5~53.0o
四、思考题
1.测定硼酸溶液时,若加NaOH液(0.1mol/L)至2.19ml时,呈现明显红色,供试品含量偏低还是高?对该制剂怎样处理才能得到合格品?
2.今有碘液,浓度为0.1095mol/L,若要配制成0.1000mol/L的碘液100ml应如何制备?
3.述5%葡萄注射液含量测定的原理、条件、并计算供试液消耗标准溶液的上下限范围。
实验十二 常用安眠镇静类药物的鉴别
一、目的:
1.掌握常用巴比妥类、吩噻嗪类药物鉴别反应的原理和方法。
2.熟悉片剂中赋形剂对鉴别试验的干扰及排除方法。
3.熟悉生物样品分析前预处理方法及TLC法在鉴别试验中的应用。
二、实验内容
(一)苯巴比妥片的鉴别:
取本品的细粉适量(约相当于苯巴比妥0.2g),加无水乙醇10ml,充分振摇,滤过,滤液置水浴上蒸干,残渣按以下方法试验。
1.丙二酰脲类的鉴别反应:
(1)取残渣约0.1g,加碳酸钠试液1ml与水10ml,振摇2分钟,滤过,滤液中逐滴加入硝酸银试液,即生成白色沉淀,振摇,沉淀即溶解;继续滴加过量的硝酸银试液,沉淀不再溶解。
(2)取残渣约50mg,加吡啶溶液(1→10)5ml,溶解后,加铜吡啶试液1ml,即生成紫色沉淀。
2.取残渣约10mg,加硫酸2滴与亚硝酸钠约5mg,混合,即显橙黄色,随即转橙红色。
3.取残渣约50mg,置试管中,加甲醛试液1ml,加热煮沸、冷却、沿管壁缓缓加硫酸0.5ml,使成两液层,置水浴中加热,接界面显玫瑰红色。
(二)注射用硫喷妥钠的鉴别:
1.取本品约0.5g,加水10ml溶解后,加过量的稀盐酸,即生产白色沉淀;滤过,沉淀用水洗净,在105℃干燥后,依法测定,溶点为157—161℃。
2.取本品约0.1g,加吡啶溶液(1→10)10ml溶解后,加铜吡啶试液1ml,振摇,放置1分钟,即生成绿色沉淀。
3.取本品约0.2g,加氢氧化钠试液5ml与醋酸铅试液2ml,生成白色沉淀;加热后,沉淀变为黑色。
4.取本品,炽灼后,显钠盐的焰色反应。
(三)尿中巴比妥类药物的鉴别:
1.对照品溶液的制备:
取硫喷妥、苯巴比妥各50mg,加甲醇5ml溶解。
2.供试品溶液的制备:
取供试尿液2ml,加盐酸(1mol/L)3滴使溶液pH值为5~6,加乙醚4ml,振摇提取约5分钟,静置,分取乙醚层,再分别加乙醚2ml同法提取2次,合并乙醚提取液,加无水硫酸钠脱水后,过滤,滤液置蒸发皿中,水浴上挥尽乙醚,残渣加甲醇数滴溶解。
3.薄层板的制备:
取硅胶GF254 2g,加0.5%羧甲基纤维素钠水溶液5.5ml调成糊状,均匀涂布于玻璃板(5×15cm)上,置水平台上晾干,在110℃活化半小时,置干燥器中备用。
4.薄层点样、展开、斑点检出:
用毛细管吸取对照液及供试液分别点于薄层板上,(原点直径约3mm),以氯仿一丙酮(9:1)为展开剂,(薄层板置缸内预饱和10分钟)展开后晾干,置紫外灯(254nm)下检视(如不明显可先熏氨后再观察)。观察结果。
(四)盐酸异丙嗪片的鉴别:
取本品,除去糖衣,研细,称取适量(约相当于盐酸异丙嗪0.2g),加水10ml,振摇使盐酸异丙嗪溶解,滤过,滤液置水浴上蒸干,残渣照以下方法试验。
1.取残渣约5mg,加硫酸5ml溶解后,溶液显樱桃红色,放置后,色渐变深。
2.取残渣约0.1g,加水3ml溶解后,加硝酸1ml,即生成红色沉淀;加热,沉淀即溶解,溶液由红色转变为橙黄色。
3.取残渣,加硝酸使成酸性后,加硝酸银试液,即生成白色凝乳状沉淀;分离,沉淀加氨试液即溶解,再加硝酸,沉淀复生成。
4.取残渣,置试管中,加等量的二氧化锰,混匀,加硫酸湿润,缓缓加热,即发生氯气。能使湿润的碘化钾淀粉试纸显蓝色。
三、说明:
1.巴比妥的银盐反应需注意碳酸钠的加入量,过多的碱与硝酸根生成沉淀,本试验以溶液中有少量未溶的巴比妥来控制碳酸钠的量。
2.TLC法鉴别巴比妥类药物也可用氯化高汞—二苯偶氮碳酰肼作显色剂,将薄板放于红外灯下烘烤至紫色背底变浅,出现显著紫色斑。该试剂对巴比妥的灵敏度为0.5~1μg,氯化高汞— 二苯偶氮碳酰肼试液的配制方法如下;
称取氯化高汞2g,二苯偶氮碳酰0.2g,分别溶于100ml甲醇中,临用前将两液等量混合。
四、思考题
1.简述上述药物鉴别反应的原理及操作注意事项。
2.如何排除片剂中赋形剂对鉴别试验的干扰?
3.尿中巴比妥的乙醚提取为何先将尿液pH值调至5~6。
实验十三 气相色谱法测定酊剂中乙醇含量
一、目的:
1.熟悉气相色谱法中内标加校正因子测定含量的方法。
2.了解气相色谱法在药物分析中的应用。
二、操作:
1.系统适用性试验:
用直径为0.25~0.18mm的二乙烯苯(为横红)乙烯苯型高分子高孔小球作为载体,柱温为120~150℃;
精密量取无水乙醇4、5、6ml,分别精密加入正丙醇5ml,加水稀释成100ml,混匀(必要时进一步稀释),取上述三份溶液适量,各进样5次,应符合下述要求:
①用正丙醇计算的塔板数应大于700;
②乙醇与正丙醇两峰的分离度应大于2;
③上述3份溶液各进样5次,所得15个校正因子变异系数不得大于2.0%。
2.标准溶液的制备
精密量取恒温至20℃的无水乙醇和正丙醇各5ml,加水稀释成100ml,混匀,即得。
3.供试溶液的制备
精密量取恒温至20℃的从试品适量(相当于乙醇约5ml)和正丙醇5ml,加水稀释成100ml,混匀,即得。
上述两种溶液必要时可进一步稀释。
4.测定法
取标准溶液与试溶液适量,分别连续注样3次,并计算出校正因子和供试品的乙醇含量,取3次计算的平均值作为结果。
三、说明:
1.在不含内标物质的供试溶液的色谱图中,内标物质峰的位置处应不出现杂质峰。
2.标准溶液与供试溶液连续3次注样所得各次校正因子和乙醇含量与其相应的平均值的相对偏差,均不得大于1.5%,否则应重新测定。
3.选用其他载体时,系统适用性验必须符合本法规定。
4.色谱柱的理论塔板数(n)计算公式为:
n=5.54(tR/Wh/2)2
其中:tR为供试品主成分或内标物质峰的保留时间,Wh/2为半高峰宽。
5.分离度(R)的计算公式为:
式中:tR2为相邻后一峰的保留时间
tR1为相邻前一峰的保留时间
W1、W2为此相邻二峰的基线峰宽
除另有规定上,分离度应大于1.5。
6.拖尾因子(T)的计算公式为:T=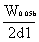

式中:W0.05h为0.05峰高处的峰宽;
d1为0.05峰高处峰极大至峰前沿之间的距离。
除另有规定外,T应在0.95~1.05间。
7.校正因子(f)的计算公式为:
式中:As为内标物质的峰面积或峰高;
Ar为对照品的峰面积或峰高;
ms为加入内标物质的量;
mr为加入对照品的量。
8.含量计算公式为:
含量(mi)=f×
式中:f为校正因子;
Ai为供试品(或其杂质)峰面积或峰高;
As为内标物质的峰面积或峰高;
ms为加入内标物质的量。
四、思考题
1.气相色谱分析中有哪几种定量方法?试简单阐述各方法的优缺点。
2.色谱系统适用试验的目的与指标是什么?
3.内标法定量时,内标的选择原则有哪些?
实验十四 血浆中苯妥英浓度的高效液
相色谱法测定
一、目的
1.熟悉用高效液相色谱法(内标法)测定血药浓度的操作;
2.练习用倍数稀释法将标准溶液稀释成工作液;
3.掌握血样中药物的萃取、浓集、萃取物重组操作以及精密移液器具的使用方法。
二、操作
1.标准溶液的配制和稀释
(1)苯妥英标准溶液:
贮备液:精密称取符合药典原料规格的苯妥钠适量,用蒸馏水溶解后,以苯妥英计再用蒸馏水定量稀释成600μg/ml准确浓度,置冰箱内4℃保存。
工作液:用5支洁净、干燥试管,照表1方法用蒸馏水稀释成a→e 5种浓度的苯妥英工作液。
表1.采用倍数稀释法配制苯妥英工作液

(2)内标(卡马西平)溶液:
精密称取符合药典原料规格的卡马西平适量,用甲醇溶解并定量稀释成200μg/ml浓度。
2.标准曲线制备
(1)标准药浓血浆的配制:
另取洁净、干燥试管5支,编号分别为A、B、C、D和E,然后照表2方法制成已知药浓的标准血浆。
表2.标准药浓血浆的配制

(2)标准血浆药浓测定方法及标准曲线建立:
于A→E各管中分别加入内标溶液0.1ml,混匀后加入0.2ml NaH2PO4缓冲液(0.1mol/L,pH7.5),混匀,加入乙酸乙酯1.5ml,置漩涡混合器上萃取10分钟后离心(2000~2500rpm)5分钟,吸取上面有机层1.2ml入另一尖底试管中,将试管置水浴中(35~40℃)并向管内液面上通入压缩空气,使乙酸乙酯萃取液缓缓挥干。于残渣中加入流动相30μl重新溶解后,进样20μl作色谱检测。用色谱数据处理机记录苯妥英和内标的峰高,以峰高比(苯妥英/内标)与对应的血浆标准药浓作标准曲线,并计算出回归方程与线性度。
测定使用的色谱条件如下:
色谱柱:C18柱(250×4.6mm,5μm)
流动相:0.01mol/L NaH2PO4(pH2.5):甲醇(50:50),用前经过滤并用超声波脱气处理。
检测器:uv254nm,灵敏度0.02AUFS
3.血浆样品中苯妥英浓度测定
取供测血浆样品0.2ml,补加0.1ml蒸馏水后完全照前述标准血浆药浓测定项下方法,自“分别加入内标溶液0.1ml……”起同法操作,最后得到供测血浆样品中的峰高比(苯妥英/内标),直接用峰高比从标准曲线查得或将其代入回归方程计算,得到血浆样品中苯妥英浓度。
三、说明
1.用苯妥英治疗血浆(或血清)浓度为10~20μg/ml,其浓度是以苯妥英计,若标准溶液是苯英钠配制,则应按分子量折算确定称量,并用苯妥英浓度表示。
2.表1稀释苯妥英标准溶液采用倍数释法,一般是临用前直接在试管内稀释。
3.表2标准曲线各管(A→E)加入的空白血浆为0.2ml,虽然加入了苯妥英工作液0.1ml使总体积变为0.3ml,但血浆样品测定当取样0.2ml后已补充了0.1ml蒸馏水,因此体积一致,故表2末栏血浆药浓以血浆0.2ml计并以每ml为单位表示,得到各管(A→E)血浆药浓分别为:30、15、7.5、3.85和1.93(μg/ml)。
4.实验中溶液的加入均采用精密移液器具(移液枪);
四、思考题
1.何谓溶剂峰和杂质峰?
如何知道杂质峰系来源于血浆基质或来源于试剂、试液?杂质峰对组份峰(药物、内标峰)有干扰时可采取哪些方法予以排除?
2.合适的内标应具备什么条件?内标加入量是否需准确知道?对内标纯度应有何要求(最低要求)?
实验十五 枸橼酸铁铵的杂质检查及
含量测定
一、目的
1.熟悉有色药物的杂质检查方法。
2.掌握碘量法在药物分析中的应用。
二、操作
1.杂质检查
氯化物 取本品0.50g,加水50ml溶解后,分取溶液10ml,加硝酸4ml,煮沸1分钟,放冷,分成2等份:1份中加硝酸银试液1ml,摇匀,放置10分钟,反复滤过,至滤液澄明,加水稀释使成约40ml后,加标准氯化钠溶液7.0ml,再加水适量使成50ml,摇匀,在暗处放置5分钟,作为对照液;另1份中加水稀释使成约40ml后,加硝酸银试液1ml,再加水适量使成50ml,摇匀,在暗处放置5分钟,如发生浑浊,与对照液比较,不得更浓。
硫酸盐 取本品0.20g,加稀盐酸17ml溶解后,煮沸1分钟,放冷,分成2等份,1份中加25%氯化钡溶液5ml,摇匀,放置10分钟,反复滤过,至滤液澄明,加水稀释使成约40ml后,加标准硫酸钾溶液5.0ml,再加水适量使成50ml,摇匀,放置10分钟,作为对照液;另1份中加水稀释使成约40ml后,加25%氯化钡溶液5ml,再加水适量使成50ml,摇匀,放置10分钟,如发生浑浊,与对照液比较,不得更浓。
铅盐 取本品0.50g,加盐酸9ml与水6ml的混合液溶解后,加硝酸0.5ml,煮沸1分钟,放冷,用乙醚振摇提取4次,每次20ml,使铁完全溶于乙醚中,如酸液仍显黄色,可再用乙醚20ml振摇提取1次,弃去乙醚液,蒸去酸液中残留的乙醚,加氨试液使成碱性后,加氰化钾试液1ml与水适量使成50ml,加硫化钠试液5滴,如显色,与对照液(取标准铅溶液1.5ml,加盐酸9ml与硝酸0.5ml,再加氨试液使成碱性后,加氰化钾试液1ml与水适量使成50ml,必要时可用稀焦糖溶液或其他无干扰的有色溶液调色后,再加硫化钠试液5滴制成)比较,不得更深。
砷盐 取本品0.40g,加水5ml溶解后,加盐酸5ml,滴加酸性氯化亚锡试液使黄色退去,加水适量使成28ml,依法检查(ChP1990版二部附录53页第一法),应符合规定。
2.含量测定
取本品约0.5g,精密称定,置具塞锥形瓶中,加水15ml溶解后,加硫酸1ml,加热至溶液由暗棕色变成淡黄色,放冷至约15℃,滴加高锰酸钾试液至溶液显粉红色并持续5秒种,加盐酸15ml与碘化钾试液15ml,密塞,静置3分钟,加水50ml稀释后,用硫代硫酸钠滴定液(0.1mol/L)滴定,至近终点时,加淀粉指示液2ml,继续滴定至蓝色消失,并将滴定的结果用空白试验校正。每1ml的硫代硫酸钠滴定液(0.1mol/L)相当于5.585mg的Fe。(本品含Fe量应为20.5~22.5%)
三、说明:
1.枸橼酸铁铵为ChP1995版以前收载品种ChP2000版以后没有再收载。
2.本品氯化物检查时采用了内消色法;滤纸应先用含硝酸的蒸馏水洗净氯化物后,方可使用。硫酸盐检查时也采用内消色法;应用含盐酸的蒸馏水将滤纸中的硫酸盐洗净后再行过滤。
实验十六 高效液相色谱法测定
阿莫西林胶囊含量
一、 目的
1. 熟悉HPLC法测定的原理及操作;
2. 掌握外标法测定阿莫西林胶囊含量。
二、 操作
1. 鉴别
取本品内容物适量,在含量测定项下记录的色谱图中,供试品主峰的保留时间应与对照品主峰的保留时间一致。
2. 含量测定
(1) 谱条件与系统适用性试验:用十八烷基硅烷键合硅胶为填充剂;以磷酸盐缓冲液(pH5.0)(取磷酸二氢钾13.6g,加水溶解后稀释到2000ml,用8mol/L氢氧化钾溶液调节pH值至5.0±0.1)-乙腈(96∶4)为流动相;流速为每分钟约1.0ml;检测波长254nm。理论板数按阿莫西林峰计算应不低于1700。
(2) 对照溶液配制:取阿莫西林对照品约30mg,精密称定,置50ml量瓶中,加磷酸盐缓冲液(pH5.0)溶解并稀释至刻度,摇匀即得。
(3) 样品测定:取装量差异项下的内容物,混合均匀,精密称取适量,加磷酸盐缓冲液(pH5.0)溶解并稀释成每ml中约含0.6mg的溶液,滤过,取续滤液20μL注入液相色谱仪,记录色谱图;另取阿莫西林对照溶液同法测定。按外标法以峰面积计算出样品中阿莫西林(C19H19N3O5S)的含量。
本品含阿莫西林(C19H19N3O5S)应为标示量的90.0%~110.0%。
三、 说明
1. 阿莫西林结构如下:
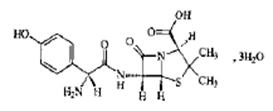

我国药典收载的相关制剂主要有片剂、胶囊剂及注射用阿莫西林钠。
附1 中华人民共和国专业标准 ZBC 10001—10007-8
药品检验操作标准原料药检验报告书
编号:
品 名 包装规格
进厂编号 厂牌来源
数 量 取样日期 年 月 日
取样件数 报告日期 年 月 日
依 据 用料车间

质检科长 复核员 检验员
附2 中华人民共和国专业标准 ZBC 10001—10007-8
药品检验操作标准成品检验报告书
编号:
品 名 包装规格
出厂编号 生产批次(检号)
数 量 取样日期 年 月 日
取样件数 报告日期 年 月 日
依 据

质检科长 复核员 检验员
附3 中华人民共和国专业标准 ZBC 10001—10007-89
药品检验操作标准
标准液配制及标定记录
名 称 配制数量 配制日期 年 月 日
基准试剂名称 标定温度 标定日期 年 月 日
指示剂名称 复标温度 复标日期 年 月 日
