光镜下显微观察蟾蜍肠系膜毛细血管,蟾蜍消化系统的解剖实习
一,实验目的:
1,观察蟾蜍肠系膜毛细血管
2,观察蟾蜍的消化系统
二,实验原理:
1,蟾蜍的正确握拿方法,动物探针的正确使用
2,蟾蜍的“双毁髓”手术技术
3,两栖类动物的解剖技术及手术剪的使用
三,动物与器材:
实验动物:雄性蟾蜍一只
实验器械:显微镜,手术剪,骨剪,镊子,吸水纸,大头针,解剖针,解剖盘,有孔的蜡板。
四, 实验步骤:
1,左手食指与中指、无名指与小指分别夹着前肢、后肢,握住蟾蜍,拇指按住吻端使头部上下活动,两耳后腺间出现一道褶线,此线中点或用金属毁髓针沿头背中线向后移动触到一凹陷处,即枕骨大孔。拇指下压使头前俯与脊柱相连处凸起,同时将毁髓针由凹陷处垂直刺入0.5~1mm,再将针从枕骨大孔向前平行刺入颅腔并在颅腔内搅动捣毁脑,然后再将毁髓针撤回至枕骨大孔,反向插入脊椎管破坏脊髓,检查蟾蜍四肢肌肉完全松驰后处死成功
2,将蟾蜍置于有孔的蛙板上,腹部朝上,四肢伸展后用大头针固定。左手用镊子提起腹部皮肤,用手术剪前端1/3处剪开一小口,并从小口处向前将腹部皮肤剪开至下颚前端。向后剪至两后肢基部之间,泄殖孔稍前方再将皮肤向两侧拉开。
3,用镊子提起腹部肌肉,用剪刀沿腹部中线剪开腹壁,拉出一段小肠,用大头针将肠系膜展开并固定在有孔的蜡板上。将肠系膜置于显微镜下观察,找到毛细血管,观察血液的流动并拍摄照片。
4,向两侧拉开蟾蜍的腹部肌肉找到蟾蜍的消化系统,小心地用剪刀和手术剪将其分离,置于解剖盘上并拍摄图片。
5,处理掉蟾蜍,洗净解剖盘和实验器械,将显微镜收好放入柜中。
五,实验结果:
1,

2,

六,实验结论:
1,毛细血管是体内分布最广、管壁最薄、口径最小的血管,仅能容纳1个红细胞通过。其管壁主要由一层内皮细胞构成,在内皮外面有一薄层结缔组织。毛细血管血流很慢,通透性大。这些特点有利于血液与组织之间充分进行物质交换。
2,蟾蜍的肝脏在体腔的前部,呈红棕色,不完全地环绕着心包,一般蛙类至少有中,右,左三叶。在中叶的腹面靠近右叶的一边有一颗黄绿色或墨绿色椭圆形的胆囊,具有导管引入胆总管而与小肠相通。
3,消化道的各个部分,在外形和构造上,都有很多变化,肺的稍后,有一个J字形的胃。胃壁富含肌肉,前端称,在之前有一段很短的食管,直接向咽腔开口。胃的后端称幽门以一圈凹隘的部分与小肠分界。小肠从幽门起弯向前方的一段,称作十二指肠,随即折向后,称为回肠,合称小肠。回肠比十二指肠长得多,经过几个曲折之后,通向一根正中宽阔的大肠。蟾蜍的大肠也就是直肠。直肠的末端下连泄殖腔。从后肢带的腹面经过,开口于体外,称为肛门。
4,脾靠近大肠的前端左侧,连附在肠系膜上,呈暗红色接近圆形。脾的着生位置在消化道的一旁,但是与消化道无关。脾并没有分泌液输送到消化道。
七,问题解答:
1,胆囊具有什么生理结构,具有什么生理功能?
胆囊分底、体、颈、管四部,颈部连胆囊管。胆囊壁由粘膜、肌层和外膜三层组成。粘膜有发达的皱襞。胆囊收缩排空时,皱襞高大而分支;胆囊充盈时,皱臂减少变矮。肝产生的胆汁经肝管排出,一般先在胆囊内贮存并浓缩,胆囊腔的容积约40~70ml。上皮细胞吸收胆汁中的水和无机盐(主要是Na+),经细胞侧面的质膜转运至上皮细胞间隙内,吸收的水和无机盐通过基膜进入固有层的血管和淋巴管内。胆囊的收缩排空受激素的调节,进食后尤其在高脂肪食物后,小肠内分泌细胞分泌胆囊收缩素,经血流至胆囊,刺激胆囊肌层收缩,排出胆汁。胆汁的作用主要是胆盐或胆汁酸的作用。胆盐或胆汁酸可作为乳化剂乳化脂肪,降低脂肪的表面张力,使脂肪乳化成微滴,分散于水溶液中,这样便增加了胰脂肪酶的作用面积。
2,肠系膜的具有什么生理功能?
蟾蜍薄而透明的肠系膜将其整个消化管悬挂在背体壁上,起到固定内脏的作用。
3,脾脏的具有什么结构,什么生理功能?
蟾蜍的脾脏与消化系统无关,而与循环系统和免疫系统有关。它是一个颜色暗红、质地柔软的网状内皮细胞器官,脾脏内部可分为红髓及白髓。红髓的主要功能是过滤和储存血液,由脾索及血窦组成,但因为其不含输入淋巴管,所以脾脏不能过滤淋巴的功能。而白髓的主要功能则为对抗外来微生物及感染。脾的组织中有许多称为“血窦”的结构,平时一部分血液滞留在血窦中,当机体失血时,血窦收缩,将这部分血液释放到外周以补充血容量。血窦的壁上附着大量巨噬细胞,可以吞噬衰老的红细胞、病原体和异物。脾脏是机体最大的免疫器官,占全身淋巴组织总量的25%,含有大量的淋巴细胞和巨噬细胞,是机体细胞免疫和体液免疫的中心。
4,蟾蜍的消化系统分别由什么结构发育而来?
内胚层演发成为消化管内侧的粘膜,以及所有由消化的突起所以形成的器官。这些器官首先形成的肝脏。它在开始呈现时,是从消化管靠近前端的腹侧,先生出一个突起,继续生长,再变成许多皱褶和分支,最后转变为一团团的细小的管子,所有的细管子皆通向总的管子,这是胆管是由最初的突起的颈部延长所成。胆管向侧壁突出,便形成胆囊。肝膨部顶端许多分支中的最内层的细胞,将来即转变成为肝的分泌细胞。至于肝中的结缔组织,血管,以及肝脏外面的被盖物等则皆则起源于中胚层。其次是胰脏,胰脏起源消化管腹侧的两个突起,最后却只形成一个器官,其导管以后才与胆管连通起来。在胰脏中只有分泌的部分以及胰管最里面的一层是起源于内胚层,至于结缔组织的血管等起源于中胚层。
5,有的同学在显微镜观察到蟾蜍体内有寄生虫,蟾蜍体内的常见的寄生虫是哪一种?
有可能是孟氏裂头绦虫。孟氏裂头绦虫又称孟氏迭宫绦虫,曼氏迭宫绦虫,是一种可以感染人畜的寄生虫。虫卵在水中发育成熟后孵出钩毛蚴,被第一中间宿主剑水蚤吞食并在其血腔中发育为原尾蚴,即第一期幼虫。剑水蚤被第二中间宿主如蝌蚪、蛇、刺猬、猪等吞食,其中主要是蛙类于蝌蚪期感染,发育为实尾蚴,即第二期幼虫,又称裂头蚴。当蝌蚪发育成蛙,裂头蚴便寄生在肌肉与内脏组织中,人生食含裂头蚴的蛙、蛇、猪等肉类可致感染。如果被猫、狗吞食,则在其小肠内发育为成虫,完成其生活史。
八,参考文献
1,《蛙体解剖学》 主编:周本湘 科学出版社 1956
2,《陈阅增普通生物学》主编:吴相钰 陈守良 葛明德 高等教育出版社 2009
3,期刊论文《胆囊组织、组织细胞膜及细胞株的FTIR研究》
- 高等学校化学学报 - 201031 (3)
4,期刊论文《保脾手术治疗外伤性脾脏破裂效果分析》
-医学信息:上旬刊-20##年 第10期 (2)
5,期刊论文《 人体孟氏裂头蚴病6例报道》
-诊断病理学杂志-20##年 第3期 (2)
第二篇:生物技术实验报告
组别:第六组
组员:苏琳伟、陈治、陈家和、陈硅
报告人:陈硅
学号:10349069
词汇&释义:
Parafilm 封口膜
Chloroform 氯仿(三氯甲烷)
Isopropanol 异丙醇
DEPC 焦碳酸二乙酯
TRIZOL a mono-phasic solution of phenol and guanidine isothiocyanate
Phenol 苯酚
isthiocyanate 异硫氰酸胍
MCS multiple cloning site (polycloning site)
注:MSC是DNA载体序列上人工合成的一段序列,含有多个限制内切酶识别位点。能为外源DNA提供多种可插入的位置或插入方案。
实验任务:
1. 从猪肌肉纤维中提取RNA;
2. 2对RNA上EGFP基因密码子片段进行RT-PCR扩增;
3. 将经RT-PCR技术扩增的cDNA与插入载体质粒,并将其转化入大肠杆菌进行克隆;
4. 从大肠杆菌中提取含目的基因的载体质粒。
实验目的: 1.掌握提取纯化总RNA的原理和技术;
2.掌握RT-PCR原理和技术;
3.掌握TA克隆原理和技术。
实验原理:
A. 总RNA提取和纯化
RNA 分离的最关键因素是尽量减少RNA 酶的污染。但RNA 酶活性非常稳定,分布广泛,除细胞内源性RNA 酶外,环境中也存在大量RNA 酶。因此在提取RNA 时,应尽量创造一个无RNA 酶的环境,包括去除外源性RNA 酶污染和抑制内源性RNA 酶活性,主要是采用焦碳酸二乙酯(DEPC)去除外源性RNA 酶,通过RNA 酶的阻抑蛋白Rnasin 和强力的蛋白质变性剂如异硫氰酸胍抑制内源性RNA 酶。
Trizol试剂是直接从细胞或组织中提取总RNA的试剂,是一种由苯酚和异硫氰酸胍组成的单相溶液它在破碎和溶解细胞时能保持RNA的完整性,裂解细胞并释放出RNA,酸性条件使RNA与DNA分离,加入氯仿后离心,样品分成水样层和有机层。RNA存在于水样层中。收集上面的的水样层后,RNA可以通过异丙醇沉淀来还原。在除去水样层后,样品中的DNA和蛋白也能相继以沉淀的方式还原。乙醇沉淀能析出中间层的DNA,在有机层中加入异丙醇能沉淀出蛋白。共纯化DNA对于样品间标准化RNA的产量十分有用。
无论是人、动物、植物还是细菌组织,各种样品最大使用量(1ml Trizol),动物组织( 50mg),植物组织(100 mg),丝状真菌( 100mg),动物细胞(5×106~1×107),酵母(1×107) 。
TRIZOL试剂能促进不同种属不同分子量大小的多种RNA的析出。例如,从大鼠肝脏抽提的RNA琼脂糖凝胶电泳并用溴化乙啶染色,可见许多介于7 kb和15 kb之间不连续的高分子量条带,(mRNA和hnRNA成分)两条优势核糖体RNA条带位于~5 kb (28S)和~2 kb (18S),低分子量RNA介于0.1 和 0.3 kb之间 (tRNA, 5S)。当抽提的RNA用TE稀释时其A260/A280比值≥1.8 。
Rnase 污染的10大来源
1:手指头 2:枪头 3:水/缓冲液4:实验台面 5:内源 Rnase 6:RNA 样品7:质粒抽提 8:RNA 保存 9:阳离子 (Ca, Mg) 10:后续实验所用的酶
RNA提取须杜绝外源酶的污染,实验时应注意
一.严格戴好口罩,手套。
二.实验所涉及的离心管,Tip 头,移液器杆,电泳槽,实验台面等要彻底处理。
三.实验所涉及的试剂/溶液,尤其是水,必须确保 RNase-Free。
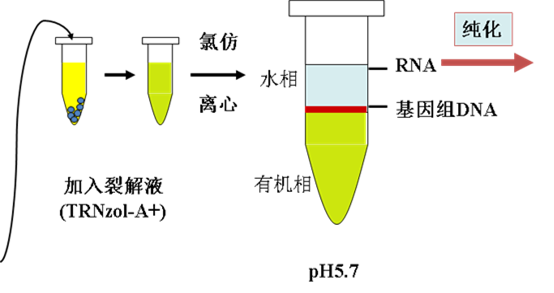
RNA纯化流程图
(附:上清不含DNA的原因:
1.因为细胞中存在DNA酶,在提取之前细胞破碎的时候没有加DNA酶抑制剂,DNA已经被分解了。
2.上层水相,PH 5.1左右,当溶液pH在酸性的时候,DNA分子就会沉淀在酚与溶液的界面,只有RNA分子留在水相。而当pH接近中性时,DNA就会溶解在水相,(导致PH中性的大概原因是trizol与样品比例不对,应该尽量保证提取量的前提下使trizol过量)
RNA纯化要求
1 纯化后不应存在对酶(如逆转录酶)有抑制作用物质
2 排除有机溶剂和金属离子的污染
3 蛋白质、多糖和脂类分子等的污染降低到最低程度
4 排除DNA分子的污染
B.RT-PCR
一、知识背景:
1、基因表达:DNA RNA Protein
单拷贝基因表达存在逐步放大机制,如一个蚕丝心蛋白基因104 个丝心蛋白mRNA(每个mRNA 存
活4d,可以合成105 个丝心蛋白) 共合成109 个丝心蛋白。因此单拷贝基因的mRNA 表达水平对
于其功能水平的调控是非常重要的。
2、PCR 技术(olymerase chain reaction):即聚合酶链式反应。
在模板、引物和四种脱氧核苷酸存在的条件下依赖于DNA 聚合酶的酶促反应,其特异性由两个人工
合成的引物序列决定。反应分三步:
a、变性:通过加热使DNA 双螺旋的氢键断裂,形成单链DNA;
b、退火:将反应混合液冷却至某一温度,使引物与模板结合。
c、延伸:在DNA 聚合酶和dNTPs 及Mg2+存在下,退火引物沿5‘ 3’方向延伸。
以上三步为一个循环,如此反复。
3、逆转录酶和RT-PCR
逆转录酶(reverse transcriptase)是存在于RNA 病毒体内的依赖RNA 的DNA 聚合酶,至少具有以下
三种活性:
1、依赖RNA 的DNA 聚合酶活性:以RNA 为模板合成cDNA 第一条链;
2、Rnase 水解活性:水解RNANA 杂合体中的RNA;
3、依赖DNA 的DNA 聚合酶活性:以第一条DNA 链为模板合成互补的双链cDNA.
二、RT-PCR 的准备:
1、引物的设计及其原则:
1)引物的特异性决定PCR 反应特异性。因此引物设计是否合理对于整个实验有着至关重要的影响。
在引物设计时要充分考虑到可能存在的同源序列,同种蛋白的不同亚型,不同的mRNA 剪切方式以及
可能存在的hnRNA 对引物的特异性的影响。尽量选择覆盖相连两个内含子的引物,或者在目的蛋白
表达过程中特异存在而在其他亚型中不存在的内含子。
2)引物设计原则的把握:引物设计原则包括
a、引物长度:一般为15~30bp ,引物太短会影响PCR 的特异性,引物太长PCR 的最适延伸温度会超
过Taq 酶的最适温度,也影响反应的特异性。
b、碱基分布:四种碱基最好应随机分布,避免嘌呤或嘧啶的聚集存在,特别是连续出现3 个以上的单
一碱基。GC 含量(Tm 值):40%~60%,PCR 扩增的复性温度一般是较低Tm 值减去5~10 度。
c、3‘端要求:3’端必须与模板严格互补,不能进行任何修饰,也不能有形成任何二级结构的可能。
末位碱基是A 时错配的引发效率最低,G、C 居中间,因此引物的3’端最好选用A、G、C 而尽可能
避免连续出现两个以上的T。
d、引物自身二级结构:引物自身不应存在互补序列,否则会自身折叠成发夹状结构或引物自身复性。
e、引物之间的二级结构:两引物之间不应有多于4 个连续碱基互补,3’端不应超过2 个。
f、同源序列:引物与非特异扩增序列的同源性应小于连续8 个的互补碱基存在。
g、5’端无严格限制:5’末端碱基可以游离,但最好是G 或C,使PCR 产物的末端结合稳定。还可
以进行特异修饰(标记、酶切位点等)等等。
根据实验目的选择适当的引物。常用引物设计软件如Primer5.0,Oligo6.0 等对于这些条件都可以自行设置。
二步法RT-PCR反应流程图示
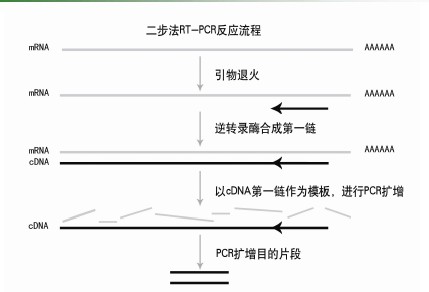
一部法RT-PCR反应流程图示
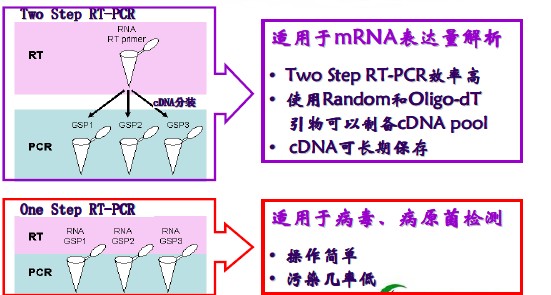
C.TA克隆
a质粒的基本特性
1.质粒的复制
通常一个质粒含有一个与相应的顺式作用控制要素结合在一起的复制起始区(整个遗传单位定义为复制子)。在不同的质粒中,复制起始区的组成方式是不同的,有的可决定复制的方式,如滚环复制和 θ 复制。在大肠杆菌中使用的大多数载体都带有一个来源于 pMB1 质粒或 ColE1 质粒的复制起始位点。图 3-1 是其复制其始示意图。
在复制时,首先合成前 RNAⅡ ,即前引物,并与 DNA 形成杂交体;而后 RNase H 切割前 RNAⅡ ,使之成为成熟的 RNAⅡ ,并形成三叶草二级结构,该引物引导质粒的复制。形成的 RNAⅠ 可控制 RNAⅡ 形成二级结构,同时 Rop 增强 RNAⅠ 的作用,从而控制质粒的拷贝数。削弱 RNAⅠ 和 RNAⅡ 之间相互作用的突变,将增加带有 pMB1 或(ColE1)复制子的拷贝数。
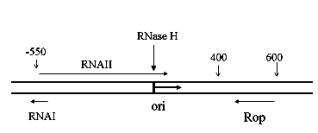
图 带 pMB1(或 ColE1) 复制起点的质粒在复制起始阶段所产生的转录的方向及其粗略大小。
滚环复制:滚环式复制(rolling circle replication)是噬菌体中常见的DNA复制方式。许多病毒DNA的复制、质粒、F因子在接合(conufgation)转移时其DNA的复制,以及许多基因扩增时都采用这种方式。
在以这种机制进行的复制中,亲代双链DNA的一条链在DNA复制起点处被切开,其5'端游离出来。这样,DNA聚合酶Ⅲ便可以将脱氧核糖核苷酸聚合在3'-OH端。当复制向前进行时,亲代DNA上被切断的5'端继续游离下来,并且很快被单链结合蛋白所结合。因为5'端从环上向下解链的同时伴有环状双链DNA环绕其轴不断的旋转,而且以3'-OH端为引物的DNA生长链则不断地以另一条环状DNA链为模板向前延伸,因而称为滚环复制。由于只有一条DNA链是完整的,因而在DNA解链时不会产生拓扑学上的问题,即未解链的双螺旋区不会产生超螺旋。当5'-端从环上解下来后不久,即与单链结合蛋白结合,以后可移动的引发体便在其上形成,以引发RNA引物的合成,然后由DNA聚合酶Ⅲ催化合成冈崎片段。这个过程与前述的DNA滞后链的合成一样。最后由DNA聚合酶Ⅰ切除RNA引物,并填充间隙构成完整的DNA链。5'端所以能从环上不断解链,主要是由于DNA聚合酶Ⅲ及引发体构成的复制体中的螺旋酶不停的向前移动所致。在这种复制方式中,DNA的延伸可以一直进行下去,产生的DNA链可以是亲代DNA单位长度的许多倍。这么长的DNA链是如何转变为单位长度的DNA分子的,目前尚不清楚。可能是由特异的内切酶切开产生单位长度的子代DNA。这些DNA可自身环绕,或保持线性分子状态。
某些质粒进行的滚环复制与噬菌体进行的滚环复制并非完全的相同,它们至少存在以下几点差别:
1.质粒在进行滚环复制时,正链和负链必须等量复制。
2.具有两个复制起始区,即双链起始区和单链起始区,它们分别起动前导链(正链)和后随链(负链)的合成。
滚环复制特点:
1、以亲本链(+链)为模板合成互补的环状负链,形成闭合环状的复制形RF1;
2、以成环滚环复制产生多个子代RF;
3、以RF的负链为模板进行滚环复制产生多拷贝正链单环。
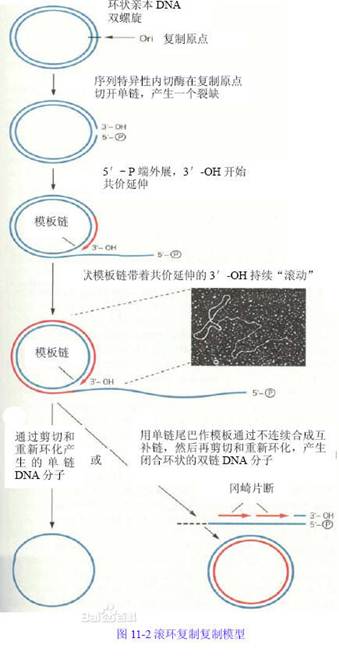
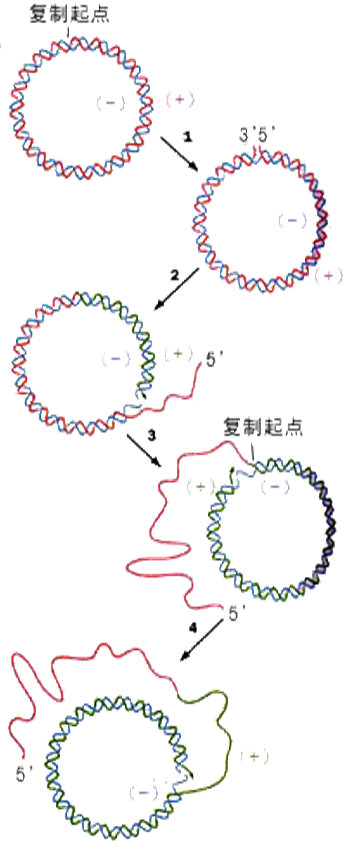

滚环复质图示
θ型复制
DNA在复制原点解开成单链状态,分别作为模板,各自合成其互补链,则出现两个叉子状的生长点,叫做复制叉。复制过程中,由于形状像希腊字母,因而叫θ复制,又叫Cairns复制,其复制中间体称为Cairns分子。在θ复制中为双向复制。大多数生物为双向等速复制,也有少数双向不等速复制的情况。
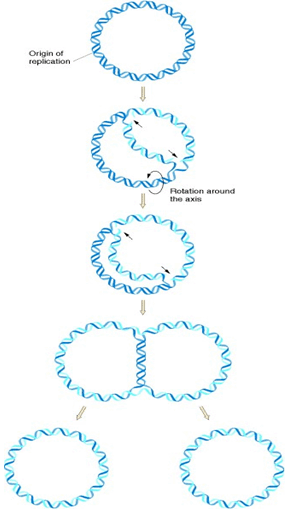
θ型复制图示
2.质粒的拷贝数
质粒拷贝数分为严谨型与松驰型。严谨型质粒每个细胞中拷贝数有限,大约 1 ~几个;松驰型质粒拷贝数较多,可达几百。表 5-1 就是不同类的质粒与复制子及拷贝数的大致关系。
表 3-1 :质粒载体及其拷贝数
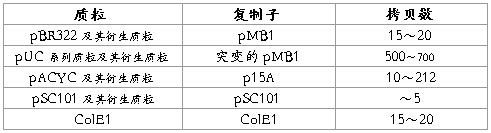
pUC 系列质粒的复制单位来自质粒 pMB1 ,但其拷贝数较高。 pMB1 质粒的复制并不需要质粒编码的功能蛋白,而是完全依靠宿主提供的半衰期较长的酶(DNA 聚合酶 Ⅰ ,DNA 聚合酶 Ⅲ),依赖于 DNA 的 RNA 聚合酶,以及宿主基因 dnaB 、 dnaC 、 dnaD 和 danZ 的产物。因此,存在抑制蛋白质合成并阻断细菌染色体复制的氯霉素或壮观霉素等抗生素时,带有 pMB1(或 ColE1) 复制子的质粒将继续复制,最后每个细胞中可积聚 2~3 千个质粒。
3.质粒的不相容性
两个质粒在同一宿主中不能共存的现象称质粒的不相容性,它是指在第二个质粒导入后,在不涉及 DNA 限制系统时出现的现象。不相容的质粒一般都利用同一复制系统,从而导致不能共存于同一宿主中。两个不相容性质粒在同一个细胞中复制时,在分配到子细胞的过程中会竞争,随机挑选,微小的差异最终被放大,从而导致在子细胞中只含有其中一种质粒。而不相容群指那些具有不相容性的质粒组成的一个群体,一般具有相同的复制子。在大肠杆菌中现已发现 30 多个不相容群,如 ColE1 和 pMB1 , pSC101 和 p15A。
4.转移性
质粒具转移性。它是指在自然条件下,很多质粒可以通过称为细菌接合的作用转移到新宿主内。它需要移动基因 mob ,转移基因 tra ,顺式因子 bom 及其内部的转移缺口位点 nic。
二、标记基因
按其用途可将标记基因分为选择标记基因和筛选标记基因。选择标记用于鉴别目标 DNA (载体)的存在,将成功转化了载体的宿主挑选出来,筛选标记可用于将特殊表型的重组子挑选出来。
(一) 选择标记
抗生素抗性基因是目前使用最广泛的选择标记。
1.氨苄青霉素抗性基因(Ampicillin resistance gene, ampr)
氨苄青霉素抗性基因是基因操作中使用最广泛的选择标记,绝大多数在大肠杆菌中克隆的质粒载体带有该基因。青霉素可抑制细胞壁肽聚糖的合成,与有关的酶结合并抑制其活性,抑制转肽反应。氨苄青霉素抗性基因编码一个酶,该酶可分泌进入细菌的周质区,抑制转肽反应并催化 β-内酰胺环水解,从而解除了氨苄青霉素的毒性。青霉素是一类化合物的总称,其分子结构由侧链 R-CO- 和主核 6-氨基青霉烷酸(6-APA)两部分组成。在 6-APA 中有一个饱和的噻唑环(A)和一个 β-内酰胺环, 6-APA 为由 L- 半脱氨酸和缬氨酸缩合成的二肽。
2.四环素抗性基因(Tetracycline resistance gene,tetr)
四环素可与核糖体 30S 亚基的一种蛋白质结合,从而抑制核糖体的转位。四环素抗性基因编码一个由 399 个氨基酸组成的膜结合蛋白,可阻止四环素进入细胞。 pBR322 质粒除了带有氨苄青霉素抗性基因外,还带有四环素抗性基因。
3.氯霉素抗性基因(chloramphenicol resistance gene, Cmr, cat)
氯霉素可与核糖体 50S 亚基结合并抑制蛋白质合成。目前使用的氯霉素抗性基因来源于转导性 P1 噬菌体(也携带 Tn9)。cat 基因编码氯霉素乙酰转移酶,一个四聚体细胞质蛋白(每个亚基 23kDa)。在乙酰辅酶 A 存在的条件下,该蛋白催化氯霉素形成氯霉素羟乙酰氧基衍生物,使之不能与核糖体结合。
4.卡那霉素和新霉素抗性基因(kanamycin/neomycin resistance gene, kanr, neor)
卡那霉素和新霉素是一种脱氧链霉胺氮基糖苷,都可与核糖体结合并抑制蛋白质合成。 卡那霉素和新霉素抗性基因实际就是一种编码氨基糖苷磷酸转移酶(APH(3')-Ⅱ, 25kDa)的基因,氨基糖苷磷酸转移酶可使这两种抗生素磷酸化,从而干扰了它们向细胞内的主动转移。在细胞中合成的这种酶可以分泌至外周质腔,保护宿主不受这些抗生素的影响。
5.琥珀突变抑制基因 supF
在基因的编码区中,若某个密码子发生突变后变成终止密码子,则称这样的突变为赭石突变(突变为 UAA),或琥珀突变(突变为 UAG),或乳白突变(突变为 UGA)。 supF 基因编码细菌的抑制性 tRNA ,可在 UAG 密码子上编译酪氨酸。如果在某一宿主中含具琥珀突变的 tetr 基因和 ampr 基因,只有当宿主含有 supF 基因时才会对 Amp 和 Tet 具有抗性。相应的, supE 基因在 UAG 密码子上编译谷氨酰氨。由于目前所用的标记基因使用方便,因此用这类标记的载体较少。
6.其它
还有一些正向选择标记,表达一种使某些宿主菌致死的基因产物,而含有外源基因片段插入后,该基因便失活。如蔗糖致死基因 SacB ,来自淀粉水解芽胞杆菌 (Bacillus amyloliquefaciens) ,编码果聚糖蔗糖酶。在含蔗糖的培养基上 sacB 基因的表达对大肠杆菌来说是致死的,因此该基因可用于插入失活筛选重组子。
(二)筛选标记
筛选标记主要用来区别重组质粒与非重组质粒,当一个外源 DNA 片段插入到一个质粒载体上时,可通过该标记来筛选插入了外源片段的质粒,即重组质粒。
1.α-互补(α-complementation)
α-互补是指 lacZ 基因上缺失近操纵基因区段的突变体与带有完整的近操纵基因区段的 β-半乳糖苷酶(β -galactosidase ,由 1024 个氨基酸组成)阴性的突变体之间实现互补。α-互补是基于在两个不同的缺陷 β-半乳糖苷酶之间可实现功能互补而建立的。大肠杆菌的乳糖 lac 操纵子中的 lacZ 基因编码 β-半乳糖苷酶,如果 lacZ 基因发生突变,则不能合成有活性的 β-半乳糖苷酶。例如, lacZ△M15 基因是缺失了编码 β-半乳糖苷酶中第 11-41 个氨基酸的 lacZ 基因,无酶学活性。对于只编码 N-端 140 个氨基酸的 lacZ 基因 (称为 lacZ') ,其产物也没有酶学活性。但这两个无酶学活性的产物混合在一起时,可恢复 β-半乳糖苷酶的活性,实现基因内互补。
在 lacZ' 编码区上游插入一小段 DNA 片段(如 51 个碱基对的多克隆位点),不影响 β-半乳糖苷酶的功能内互补。但是,若在该 DNA 小片段中再插入一个片段,将几乎不可避免地导致产生无α-互补能力的 β-半乳糖苷酶片段。利用这一互补性质,可用于筛选在载体上插入了外源片段的重组质粒。在相应的载体系统中,lacZ△M15 放在 F 质粒上, 随宿主传代; lacZ' 放在载体上, 作为筛选标记 (图 3-2) 。相应的受体菌有 JM 系列、 TG1 和 XL1-Blue ,前二者均带有 D (lac - proAB)F'[ proAB + lacIq lacZD M15] 基因型。其中 lacI 为 lac 阻抑物的编码基因,lacIq 突变使阻抑物产量增加,防止 lacZ 基因渗漏表达。
lacZ 基因是乳糖 lac 操纵子中编码 β-半乳糖苷酶的基因,乳糖及其衍生物可诱导其表达。 乳糖既是 lac 操纵子的诱导物,也是作用的底物。异丙基-β-D- 硫代半乳糖苷(IPTG)是乳糖的衍生物,可作为 lac 操纵子的诱导物,但不能作为反应的底物; 5-溴-4-氯-3-吲哚-β-D-半乳糖苷(X-gal) 可作为 lac 操纵子的底物,但不能作为诱导物。底物 X-gal 还可充作生色剂,被 β-半乳糖苷酶分解后可产生兰色产物,可使菌落或噬菌斑呈兰色。
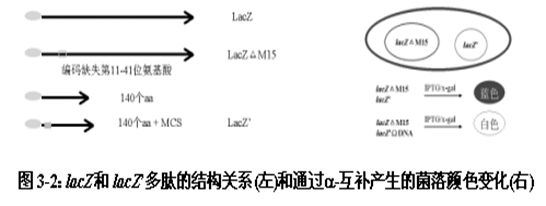
2.插入失活
通过插入失活进行筛选的质粒主要有 pBR322 ,该质粒具有四环素抗性基因(tetr)和氨苄青霉素抗性基因(ampr)两种抗性标记。当外源 DNA 片段插入 tetr 基因后,导致 tetr 基因失活,变成只对氨苄青霉素有抗性。这样就可通过对抗生素是双抗还是单抗来筛选是否有外源片段插入到载体中。
三、质粒载体的种类
(一)克隆载体
克隆载体主要用于扩增或保存 DNA 片段,是最简单的载体。
1.pBR322
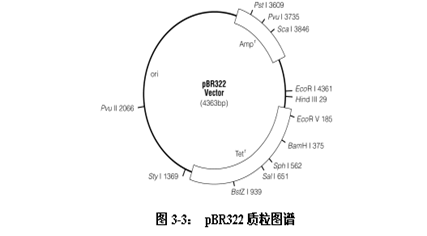
pBR322 质粒的大小为 4361bp , GenBank 注册号为 V0lll9 和 J01749 ,含有 30 多个单一位点,具有四环素抗性基因(tetr)和氨苄青霉素抗性基因(ampr),其质粒复制区来自 pMB1 (如图 3-3)。目前使用广泛的多质粒载体几乎都是由此发展而来的。利用四环素抗性基因内部的 BamHⅠ 位点来插入外源 DNA 片段,可通过插入失活进行筛选。
2.pUC18 和 pUC19
pUC18 和 pUC19 大小只有 2686bp ,是最常用的质粒载体,其结构组成紧凑,几乎不含多余的 DNA 片段,GenBank注册号为 L08752(pUC18)和 X02514(pUC19)。由 pBR322 改造而来,其中 lacZ (MSC) 来自 M13mp18/19 图 3-4 是其质粒图谱。
这两个质粒的结构几乎是完全一样 的,只是多克隆位点的排列方向相反。这些质粒缺乏控制拷贝数的 rop 基因,因此其拷贝数达 500-700 。 pUC 系列载体含有一段 lacZ 蛋白氨基末端的部分编码序列,在特定的受体细胞中可表现 α-互补作用。因此在多克隆位点中插入了外源片段后,可通过 α-互补作用形成的蓝色和白色菌落筛选重组质粒。

图 3-4 : pUC18/19 质粒图谱
3.pUC118 和 pUC 119
由 pUC18/19 增加了一些功能片段改造而来,大小为 3162bp , GenBank 注册号为 U07649(pUC118)和 U07650(pUC119)。相当于在 pUC18/19 中增加了带有 M13 噬菌体 DNA 合成的起始与终止以及包装进入噬菌体颗粒所必需的顺式序列。
4.pGEM-3Z/4Z
pGEM-3Z/4Z由pUC18/19 增加了一些功能片段改造而来,大小为 2.74kb, GenBank 注册号为 X65304(pGEM-3Z, 2743bp)和 X65305(pGEM-4Z, 2746)。与 pUC18/19 相比,在多克隆位点的两端添加了噬菌体的转录启动子,如 Sp6 和 T7 噬菌体的启动子。 pGEM-3Z 和 pGEM-4Z 的差别在于二者互换了两个启动子的位置。
5.多功能质粒载体
在上述载体的基础上,人们设计出一些多功能的质粒载体,这类质粒载体综合了以上质粒的特点。除了作为质粒载体基本要素外,综合了上述功能要素,如多克隆位点、 α-互补、噬菌体启动子和单链噬菌体的复制与包装信号。典型的这类质粒有 pBluescriptⅡKS(±),这类质粒一般由 4 个质粒组成一套系统,其差别在于多克隆位点方向相反(根据多克隆位点两端 KpnⅠ 和 SacⅠ 的顺序,用 KS 或 SK 表示),或单链噬菌体的复制启始方向相反(或者说,引导 DNA 双链中不同链合成单链 DNA ,用 + 或 - 表示), pBluescriptⅡKS(+) 的 GenBank注册号为 X52327 。 pBluescriptⅡKS(±) 的多克隆位点与 pUC18/19 的不同,且使用 f1 噬菌体的复制与包装信号序列,质粒图谱如图 3-5 。

6.表达载体
该类载体是在常规克隆载体的基础上衍生而来的,主要增添了强启动子,以及有利于表达产物分泌、分离或纯化的元件,
有关本次实验使用的
b.本次实验所用pGEM®-T Easy 载体的有关信息
载体图谱 pGEM®-T 载体和pGEM®-T Easy 载体多克隆位点序列
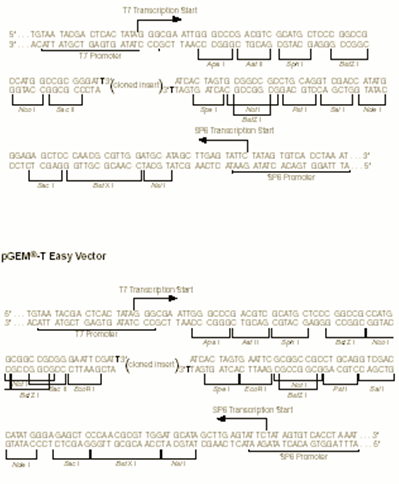
pGEM®-T Easy 载体图和相关序列位点
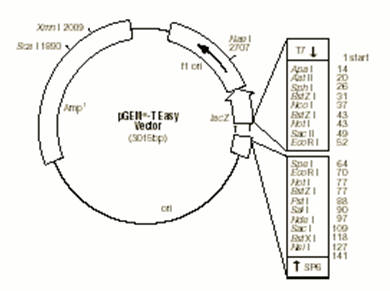
pGEM®-T Easy 载体相关的序列位点
T7 RNA 聚合酶转录起始位点 1
多克隆位点区 10-128
SP6 RNA 聚合酶启动子(-17 至+3) 139-158
SP6 RNA 聚合酶转录起始位点 141
pUC/M13 反向测序引物结合位点 176-197
lacZ 起始密码子 180
lac 操纵子 200-216
β内酰胺酶编码区 1337-2197
噬菌体f1 区 2380-2835
lac 操作子序列 2836-2996, 166-395
pUC/M13 正向测序引物结合位点 2956-2972
T7 RNA 聚合酶启动子(-17 至+3) 2999-3
pGEM®-T 和pGEM®-T Easy 载体的特殊应用
1.PCR 产物的克隆。
2.采用Erase-a-Base®系统构建不定向巢式缺失体。
3.单链DNA 制备。
4.重组子的蓝白斑筛选。
5.利用双向启动子进行体外转录。
注意平端PCR 产物(来自质粒载体说明书)
具有校正活性的热稳定性DNA 聚合酶如Pfu DNA 聚合酶 (产品目录号:M7741),
Pwo DNA 聚合酶,Tli DNA 聚合酶(目录号M7101)在进行PCR 扩增时产生平端PCR
产物。这种平端PCR 产物可以通过加A 尾程序后(图4),连接到pGEM®-T 和pGEM®-T
Easy 载体中(6)。采用这种方法连接,只有一个片段插入,而平端连接克隆可能产生
多个插入。另外采用T 载体克隆不需要对载体去磷酸化,载体自连的背景很低。
Pfu 和Tli DNA 聚合酶产生的PCR 产物经过加尾处理,并以理想载体:插入片段
比例进行连接,可得到55-95%重组子(表2)。值得注意的是,PCR 产物有必要采
用Wizard® SV Gel and PCR Clean-Up System(d) (Cat.# A9281)进行PCR 产物直接纯
化或凝胶纯化。若PCR 产物不进行纯化,Pfu、Pwo 和Tli DNA 聚合酶的校正活性在
进行加尾反应或连接反应时,可降解PCR 片段的3’-A 或除去加尾反应的载体的3’-T
突出端。
为了提高克隆效率,应根据纯化PCR 产物的摩尔浓度调整加尾反应中的DNA 量
和连接需要的体积。当PCR 产物片段小或扩增反应良好,产物的摩尔浓度高,加尾或
连接反应需要的体积很小。相反,如果PCR 片段比较大或扩增不好,产物的摩尔浓度
低,则需要较大的体积。我们已成功地用Taq DNA 聚合酶 对1-7μl 纯化的平端PCR
片段进行加尾反应,并优化插入片段:载体的连接比例,参见章节IV.C 详细讨论了插入
片段:载体比例的优化。转化后采用蓝白斑筛选重组子,结果70-100%的重组子含有
PCR 扩增的DNA 片段,而PCR 片段没有加尾的对照反应,只有很少的重组子。对照
实验结果表明大多数pGEM®-T Easy 载体含有3’-T,并且Taq DNA 聚合酶可成功对大
多数PCR 片段加上3’-A。
优化插入片段和载体的摩尔比
pGEM®-T 和pGEM®-T Easy 载体系统优化的插入DNA 对照片段和载体的摩尔比为1:1,采用8:1 到1:8 的连接比例也可成功进行连接。如果你的PCR 产物开始的连接不理想,则有必要优化连接比例。一般开始采用3:1 到1:3 的连接比例。
PCR 产物的浓度可通过凝胶电泳上的DNA 分子量标准进行估计或采用荧光定量。pGEM®-T 和pGEM®-T Easy 载体大概3kb 大小,系统提供的载体浓度为50ng/μl。
计算连接反应中需要的PCR 产物的量可采用以下公式:
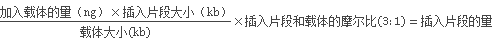
根据推荐的不同插入片段:载体比例加入足够的pGEM®-T 和pGEM®-T Easy载体进行连接,并进行对照反应。
c 重组T质粒的构建
外源DNA与载体分子的连接就是DNA重组,这样重新组合的DNA叫做重组体或重组子。重组的DNA分子是在DNA连接酶的作用下,有Mg2+、ATP存在的连接缓冲系统中,将分别经酶切的载体分子与外源DNA分子进行连接。DNA连接酶有两种:T4噬菌体DNA连接酶和大肠杆菌DNA连接酶。两种DNA连接酶都有将两个带有相同粘性末端的DNA分子连在一起的功能,而且T4噬菌体DNA连接酶还有一种大肠杆菌DNA连接酶没有的特性,即能使两个平末端的双链DNA分子连接起来。但这种连接的效率比粘性末端的连接率低,一般可通过提高T4噬菌体DNA连接酶浓度或增加DNA浓度来提高平末端的连接效率。T4噬菌体DNA 连接酶催化DNA 连接反应分为3 步:首先,T4 DNA 连接酶与辅因子ATP形成酶-ATP复合物;然后,酶-ATP复合物再结合到具有5’磷酸基和3’羟基切口的DNA上,使DNA腺苷化;最后产生一个新的磷酸二酯键,把切口封起来。连接反应通常将两个不同大小的片断相连。因为DNA片断有两个端点,所以切割时出现两种可能,一种是单酶切,另一种是双酶切,这两种酶切方法在基因工程操作中都经常用到。对于单酶切来说,载体与供体的末端都相同,连接可以在任何末端之间进行,这样就导致了大量的自连接产物。为了减少自环的高本底,可对载体进行5’除磷酸处理,原理是连接酶只能连接DNA片断的3’OH末端与5’端,所以除磷后载体不会自环。一旦有外源片断插入时,由外源片断提供5’端就能与载体进行连接。通过这种方法可大大减少由载体的自 环造成的高本底。对于双酶切来说,无论载体与供体同一片段上都有不同的末端,这样就避免了载体与供体的自环,能使有效连接产物大大增加。双酶切的另一个特点是能将供体分子定向连接到载体上。 连接反应的温度在37℃时有利于连接酶的活性。但是在这个温度下粘末端的氢键结合是不稳定的。因此采取折中的温度,即12-16℃,连接12-16h(过夜),这样既可最大限度地发挥连接酶的活性,又兼顾到短暂配对结构的稳定。Taq DNA酶扩增的PCR产物,其DNA双链前后末端都有一个游离的A碱基,可以与pGEM-T Easy Vector 末端游离的T碱基互补形成环状重组T质粒。
d 质粒转化与筛选
(1) 感受态细胞(Competent cells):受体细胞经过一些特殊方法(如CaCl,RuCl等化学试剂法)的处理后,细胞膜的通透性发生变化,成为能容许外源DNA的载体分子通过,进入感受态细胞
(2) 转化(transformation):是将异源DNA分子引入一细胞株系,使受体细胞获得新的遗传性状的一种手段,是基因工程等研究领域的基本实验技术。
注意: 转化过程所用的受体细胞一般是限制-修饰系统缺陷的变异株,即不含限制性内切酶和甲基化酶的突变株。
(3) 转化的方法:
i化学方法(热击法):使用化学试剂(如CaCl2、RuCl等)制备的感受态细胞,通过热击处理将载体分子导入受体细胞;
ii电转化法:使用低盐缓冲液或水洗制备的感受态细胞,通过高压麻脉冲的作用将载体DNA分子导入受体细胞。
pGEM®-T 和pGEM®-T Easy 载体连接反应产物的转化步骤
一定要使用高效率的感受态细胞(1×108 cfu/μg DNA)进行转化,因为采用单碱
基突出端进行连接不是很有效,采用转化效率高的感受态细胞1×108 cfu/μg DNA(或
更高)可得到合理的克隆菌数目(参见章节VI. E)。我们建议使用JM109 高效感受态细胞(产品目录号:L2001),pGEM®-T 和pGEM®-T Easy 载体系统Ⅱ提供这种感受态细胞。也可使用其他宿主菌,但是它们应该适合用蓝白斑及氨苄进行筛选。在使用JM109 制备感受态细胞前,JM109 应保存在含加维生素B1 的M9 基本培养基中,这有助于F’因子的选择,F’因子带有proAB 基因,这和脯氨酸营养缺陷型宿主菌(proAB 基因缺失)是互补的,携带的lacIqZΔM15 对于蓝白斑筛选是必需的。如果你不采用Promega 公司JM109 高效率感受态细胞,应按照后面的转化步骤进行操作。在LB/氨苄/IPTG/X-Gal 平板筛选转化子(参见章节XI. A),为得到理想的结果,最好不要使用放置超过1 个月的平板。
筛选含插入片段的转化子
插入片段成功克隆到pGEM®-T 和pGEM®-T Easy 载体中,可阻断β 半乳糖苷酶
的编码序列,因而重组克隆可在指示培养基上通过颜色进行筛选。然而连接到pGEM®-T
和pGEM®-T Easy 载体的PCR 产物的特点对转化后蓝白斑克隆菌的比例影响很大。大
多数情况下含有PCR 产物的克隆菌为白色,如果PCR 片段和LacZ 基因在同一读码框,
则重组克隆菌可能为蓝色。这种DNA 片段碱基数是3 的整数倍(含3’-A),并且读码
框无终止密码子。据文献报道,在同一读码框内插入长达2kb 的DNA 片断,重组克隆
菌仍可能为蓝色。
即使PCR 产物不是3 的整数倍大小,由于扩增反应可引入突变(缺失或点突变),
也可造成pGEM®-T 和pGEM®-T Easy 载体和片段连接转化后,重组克隆菌为蓝色。
pGEM®-T 和pGEM®-T Easy 载体系统的对照DNA 为542bp,来源于Promega
公司pGEM®-luc(b,个g)DNA (Cat.#E1541),这个DNA 序列被引入突变,在6 个读码框
中含有多个终止密码子,从而确保对照反应产生的蓝色克隆菌的背景数很低。由此可见,
插入DNA 对照的结果不能代表你的PCR 产物的连接结果。(这就解释了为什么对菌落进行插入片段进行PCR扩增后进凝胶电泳分析是必要的。)
E. 质粒提取
细菌质粒是一类双链、闭环的DNA,大小范围从1kb至200kb以上不等。各种质粒都是存在于细胞质中、独立于细胞染 色体之外的自主复制的遗传成份,通常情况下可持续稳定地处于染色体外的游离状态,但在一定条件下也会可逆地整合到寄主染色体上,随着染色体的复制而复制, 并通过细胞分裂传递到后代。
质粒已成为目前最常用的基因克隆的载体分子,重要的条件是可获得大量纯化的质粒DNA分子。目前已有许多方法可用于质 粒DNA的提取,本实验采用碱裂解法提取质粒DNA。
碱裂解法是一种应用最为广泛的制备质粒DNA的方法,其基本原理为:当菌体在NaOH和 SDS溶液中裂解时,蛋白质与DNA发生变性,当加入中和液后,质粒DNA分子能够迅速复性,呈溶解状态,离心时留在上清中;蛋白质与染色体DNA不变性 而呈絮状,离心时可沉淀下来。
纯化质粒DNA的方法通常是利用了质粒DNA相对较小及共价闭环两个性质。例如,氯化铯-溴化乙锭梯度平衡离心、离 子交换层析、凝胶过滤层析、聚乙二醇分级沉淀等方法,但这些方法相对昂贵或费时。对于小量制备的质粒DNA,经过苯酚、氯仿抽提,RNA酶消化和乙醇沉淀 等简单步骤去除残余蛋白质和RNA,所得纯化的质粒DNA已可满足细菌转化、DNA片段的分离和酶切、常规亚克隆及探针标记等要求,故在分子生物学实验室 中常用。
一、试剂准备
1. 溶液Ⅰ: 50mM葡萄糖,25mM Tris-HCl(pH 8.0),10mM EDTA(pH 8.0)。1M Tris-HCl (pH 8.0)12.5ml,0.5M EDTA(pH 8.0)10ml,葡萄糖4.730g,加ddH2O至500ml。在10 lbf/in2高压灭菌15min ,贮存于4℃。
任何生物化学反应,首先要控制好溶液的pH,因此用适当浓度的和适当pH值的Tris-Cl溶液。50 mM葡萄糖最大的好处只是悬浮后的大肠杆菌不会快速沉积到管子的底部。因此,如果溶液I中缺了葡萄糖其实对质粒的抽提本身而言,几乎没有任何影响。所以说溶液I中葡萄糖是可缺的。EDTA呢?大家知道EDTA是Ca2+和Mg2+等二价金属离子的螯合剂,配在分子生物学试剂中的主要作用是:抑制DNase的活性,和抑制微生物生长。在溶液I中加入高达 10 mM 的EDTA,无非就是要把大肠杆菌细胞中的所有二价金属离子都螯合掉。如果不加EDTA,其实也没什么大不了的,只要不磨洋工,只要是在不太长的时间里完 成质粒抽提,就不用怕DNA会迅速被降解,因为最终溶解质粒的TE缓冲液中有EDTA。如果哪天你手上正好缺了溶液I,可不可以抽提质粒呢?实话告诉你, 只要用等体积的水,或LB培养基来悬浮菌体就可以了。
NaOH也使DNA变性,但只是个副产物,在溶液3加入后其中的醋酸和NaOH中和,质粒DNA恢复活性
2. 溶液Ⅱ:0.2N NaOH,1% SDS。2N NaOH 1ml,10%SDS 1ml,加ddH2O至10ml。使用前临时配置 。
这是用新鲜的0.4 N的NaOH和2%的SDS等体积混合后使用的。要新从浓NaOH稀释制备0.4N的NaOH,无非是为了保证NaOH没有吸收空气中的CO2而减弱了碱 性。很多人不知道其实破细胞的主要是碱,而不是SDS,所以才叫碱法抽提。事实上NaOH是最佳的溶解细胞的试剂,不管是大肠杆菌还是哺乳动物细胞,碰到了碱都会几乎在瞬间就溶解,这是由于细胞膜发生了从bilayer(双层膜)结构向 micelle(微囊)结构的相变化所导致。用了不新鲜的0.4 N NaOH,即便是有SDS也无法有效溶解大肠杆菌(不妨可以自己试一下),自然就难高效率抽提得到质粒。如果只用SDS当然也能抽提得到少量质粒,因为 SDS也是碱性的,只是弱了点而已。很多人对NaOH的作用误以为是为了让基因组DNA变性,以便沉淀,这是由于没有正确理解一些书上的有关DNA变性复 性的描述所导致。有人不禁要问,既然是NaOH溶解的细胞,那为什么要加SDS呢?那是为下一步操作做的铺垫。这一步要记住两点:第一,时间不能过长,千 万不要这时候去接电话,因为在这样的碱性条件下基因组DNA片断会慢慢断裂;第二,必须温柔混合(象对待女孩子一样),不然基因组DNA也会断裂。基因组 DNA的断裂会带来麻烦。
3. 溶液Ⅲ:醋酸钾(KAc)缓冲液,pH 4.8。5M KAc 300ml,冰醋酸 57.5ml,加ddH2O至500ml。4℃保存备用。
溶液III加入后就会有大量的沉淀,但大部分人却不明白这沉淀的本质。最容易产生的误解是,当SDS碰到酸性后发生的沉淀。如果你这样怀疑,往1%的 SDS溶液中加如2M的醋酸溶液看看就知道不是这么回事了。大量沉淀的出现,显然与SDS的加入有关系。如果在溶液II中不加SDS会怎样呢,也会有少量 的沉淀,但量上要少得多,显然是盐析和酸变性沉淀出来的蛋白质。既然SDS不是遇酸发生的沉淀,那会不会是遇盐发生的沉淀呢?在1%的SDS溶液中慢慢加 入5 N的NaCl,你会发现SDS在高盐浓度下是会产生沉淀的。因此高浓度的盐导致了SDS的沉淀。但如果你加入的不是NaCl而是KCl,你会发现沉淀的量 要多的多。这其实是十二烷基硫酸钠(sodium dodecylsulfate)遇到钾离子后变成了十二烷基硫酸钾(potassium dodecylsulfate, PDS),而PDS是水不溶的,因此发生了沉淀。如此看来,溶液III加入后的沉淀实际上是钾离子置换了SDS中的钠离子形成了不溶性的PDS,而高浓度 的盐,使得沉淀更完全。大家知道SDS专门喜欢和蛋白质结合,平均两个氨基酸上结合一个SDS分子,钾钠离子置换所产生的大量沉淀自然就将绝大部分蛋白质 沉淀了,让人高兴的是大肠杆菌的基因组DNA也一起被共沉淀了。这个过程不难想象,因为基因组DNA太长了,长长的DNA自然容易被SDS给共沉淀了,尽管SDS并不与DNA分子结合。
那么2 M的醋酸又是为什么而加的呢?是为了中和NaOH,因为长时间的碱性条件会打断DNA,所以要中和之。基因组DNA一旦发生断裂,只要是50-100 kb大小的片断,就没有办法再被PDS共沉淀了。所以碱处理的时间要短,而且不得激烈振荡,不然最后得到的质粒上总会有大量的基因组DNA混入,琼脂糖电 泳可以观察到一条浓浓的总DNA条带。很多人误认为是溶液III加入后基因组DNA无法快速复性就被沉淀了,这是天大的误会,因为变性的也好复性的也 好,DNA分子在中性溶液中都是溶解的。NaOH本来是为了溶解细胞而用的,DNA分子的变性其实是个副产物,与它是不是沉淀下来其实没有关系。溶液 III加入并混合均匀后在冰上放置,目的是为了PDS沉淀更充分一点。
4. TE:10mM Tris-HCl(pH 8.0),1mM EDTA(pH 8.0)。1M Tris-HCl(pH 8.0)1ml,0.5M EDTA(pH 8.0)0.2ml,加ddH2O至100ml。15 lbf/in2高压湿热灭菌20min,4℃保存备用。
5.苯酚/氯仿 /异戊醇(25:24:1)
不要以为PDS沉淀的形成就能将所有的蛋白质沉淀了,其实还有很多蛋白质不能被沉淀,因此要用酚/氯仿/异戊醇进行抽提,然后进行酒精沉淀才能得到质量稳 定的质粒DNA,不然时间一长就会因为混入的DNase而发生降解。这里用25/24/1的酚/氯仿/异戊醇是有很多道理的,这里做个全面的介绍。酚 (Phenol)对蛋白质的变性作用远大于氯仿,按道理应该用酚来最大程度将蛋白质抽提掉,但是水饱和酚的比重略比水重,碰到高浓度的盐溶液(比如4M的 异硫氰酸胍),离心后酚相会跑到上层,不利于含质粒的水相的回收;但加入氯仿后可以增加比重,使得酚/氯仿始终在下层,方便水相的回收;还有一点,酚与水 有很大的互溶性,如果单独用酚抽提后会有大量的酚溶解到水相中,而酚会抑制很多酶反应(比如限制性酶切反应),因此如果单独用酚抽提后一定要用氯仿抽提一 次将水相中的酚去除,而用酚/氯仿的混合液进行抽提,跑到水相中的酚则少得多,微量的酚在乙醇沉淀时就会被除干净而不必担心酶切等反应不能正常进行。至于 异戊醇的添加,其作用主要是为了让离心后上下层的界面更加清晰,也方便了水相的回收。
6.乙醇(无水乙醇、70%乙醇)
回收后的水相含有足够多的盐,因此只要加入2倍体积的乙醇,在室温放置几分钟后离心就可以将质粒DNA沉淀出来。这时候如果放到-20℃,时间一长反而会 导致大量盐的沉淀,这点不同于普通的DNA酒精沉淀回收,所以不要过分小心了。高浓度的盐会水合大量的水分子,因此DNA分子之间就容易形成氢键而发生沉 淀。如果感觉发生了盐的沉淀,就用70%的乙醇多洗几次,每次在室温放置一个小时以上,并用tip将沉淀打碎,就能得到好的样品。得到的质粒样品一般用含 RNase(50 ug/ml)的TE缓冲液进行溶解,不然大量未降解的RNA会干扰电泳结果的.
Q&A
(1)为什么用无水乙醇沉淀DNA?
用无水乙醇沉淀DNA,这是实验中最常用的沉淀DNA的方法。乙醇的优点是可以任意比和水相混溶,乙醇与核酸不会起任何化学反应,对DNA很安全,因此是理想的沉淀剂。 DNA溶液是DNA以水合状态稳定存在,当加入乙醇时,乙醇会夺去DNA周围的水分子,使DNA失水而易于聚合。一般实验中,是加2倍体积的无水乙醇与DNA相混合,其乙醇的最终含量占67%左右。因而也可改用95%乙醇来替代无水乙醇(因为无水乙醇的价格远远比95%乙醇昂贵)。但是加95%的乙醇使总体积增大,而DNA在溶液中有一定程度的溶解,因而DNA损失也增大,尤其用多次乙醇沉淀时,就会影响收得率。折中的做法是初次沉淀DNA时可用95%乙醇代替无水乙酵,最后的沉淀步骤要使用无水乙醇。也可以用0.6倍体积的异丙醇选择性沉淀DNA。一般在室温下放置15-30分钟即可。
( 2) 在用乙醇沉淀DNA时,为什么一定要加NaAc或NaCl至最终浓度达0.1~0.25mol/L?
在pH为8左右的溶液中,DNA分子是带负电荷的,加一定浓度的NaAc或NaCl,使Na+中和DNA分子上的负电荷,减少DNA分子之间的同性电荷相斥力,易于互相聚合而形成DNA钠盐沉淀,当加入的盐溶液浓度太低时,只有部分DNA形成DNA钠盐而聚合,这样就造成DNA沉淀不完全,当加入的盐溶液浓度太高时,其效果也不好。在沉淀的DNA中,由于过多的盐杂质存在,影响DNA的酶切等反应,必须要进行洗涤或重沉淀。
三.实验内容
Part I. 总RNA提取
【材料与试剂】
l 转基因猪肌肉组织
l TRIZOL溶液
l 氯仿
l 异丙醇
l DEPC H2O
l 75% EtOH
【方法与步骤】
Part IA 提取总RNA
组织提取
1. 从液氮中取出用锡纸包裹的转基因小鼠肌肉组织0.6 g,在液氮中将组织研磨成粉末,液氮挥发完全后加入TRIZOL试剂4.5 ml,混匀并转移到1.5 ml离心管,每管约装1ml(装四管)
2. 在室温下静置5 min。
分相
3. 按每1mlTRIZOl加入0.2 ml氯仿,震荡混匀15 s. 在室温静置10 min,此时溶液应该分相3层。
4. 12000 rpm 离心15 min。上层:RNA(约为Trizol的60%);中间:DNA; 下层:蛋白质(酚-氯仿)。
5. 吸取0.4ml上清液于另一EP管中。
RNA沉淀
6. 在上清液中按0.4 ml/1 mlTRIZOl的量加入异丙醇,混合后在室温下静止10 min
7. 离心 12000 rpm 10分钟4℃。
8. 弃上清。
RNA清洗
9. 按最初匀浆时加入的TRIZOL量每1mlTRIZOL加入 1 ml 75% EtOH,震荡均匀。
10. 4℃ 8000 rmg 10 min。
11. 弃上清并在4℃ 12000 rmp离心5 min,弃上清。
12. 倒置离心管于滤纸上,在空气中干燥沉淀,但不能完全干燥(20-30 min)。
13. 每管沉淀用20-25ul DEPC反复吹打溶解,在60℃下温育5 min。
14. 每管取10μl 于另一离心管中。剩余样品在-80℃下保存(避免反复冻融)。
Part IB RNA检测与定量
1. 分别测量样品的A260nm和A260nm与A280nm的比值,通过OD值得到样品RNA的浓度及纯度。(分离RNA的理想效果是A260/A280=1.8~2.0)。
2. 取1μgRNA进行琼脂糖凝胶电泳。
Part II. RT-PCR
【材料与试剂】
l PrimeScript RT-PCR Kit (Takara, DRR014, 50 reations)
l EGFP-F primer
l EGFP-R primer
【方法与步骤】
Part IIA 反转录反应
1. 按以下体系配置溶液(另设NTC对照一管不加总RNA)

2. 将反应体系在65℃下加热5 min 后立即置于冰上。
3. 混匀溶液,并按下表配置反转录反应体系。

4. 将反应体系在42℃下加热30 min,然后95℃下加热5 min, 后置于冰上。
Part IIB. PCR扩增
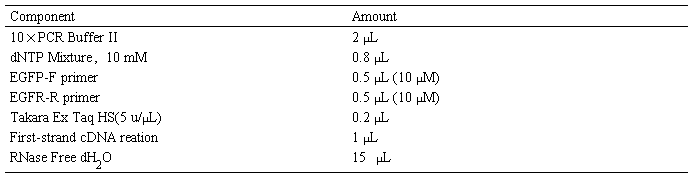
1. 按下表配置20 μL PCR反应体系。
3. 进行PCR反应,参数设置如下:
预变性:95℃ 5 min
35个循环(变性;引物退火;引物延申):94℃ 30 sec;58℃ 30 sec;72℃ 30 sec
最后延伸:72℃ 5min。
4. 对PCR产物进行凝胶电泳分析
Part III. TA克隆
Part IIIA. 回收含有目的基因的胶
【材料与试剂】
Axygen® AxyPrep? DNA Gel Extration Kit
【步骤与方法】
1. 在紫外灯下切下含有目的DNA 的琼脂糖凝胶,用纸巾吸尽凝胶表面液体并切碎。计算凝胶重量(提前记录1.5 ml 离心管重量),该重量作为一个凝胶体积(如100 mg=100 μl 体积)。
2. 加入3 个凝胶体积的Buffer DE-A,混合均匀后于75°C 加热(低熔点琼脂糖凝胶于40°C 加热),间断混合(每2-3 min),直至凝胶块完全熔化(约6-8 min)。
* Buffer DE-A 为红色溶液。在熔化凝胶的过程中,可以帮助观察凝胶是否完全熔化。
3. 加0.5 个Buffer DE-A 体积的Buffer DE-B,混合均匀。当分离的DNA 片段小于400bp 时,需再加入1 个凝胶体积的异丙醇。
* 加Buffer DE-B 后混合物颜色变为黄色,充分混匀以保证形成均一的黄色溶液。
步骤4-6 可以选择负压法或离心法。
4. 吸取步骤3 中的混合液,转移到DNA 制备管(置于2 ml(试剂盒内提供)离心管)中,12,000×g
离心1 min。弃滤液。
5. 将制备管置回2 ml 离心管,加500 μl Buffer W1,12,000×g 离心30 s,弃滤液。
6. 将制备管置回2 ml 离心管,加700 μl Buffer W2,12,000×g 离心30 s,弃滤液。以同样的方法再用700 μl Buffer W2 洗涤一次12,000×g 离心1 min。
* 确认在Buffer W2 concentrate 中已按试剂瓶上的指定体积加入无水乙醇。
* 两次使用Buffer W2 冲洗能确保盐份被完全清除,消除对后续实验的影响。
7. 将制备管置回2 ml 离心管中,12,000×g 离心1 min。
8. 将制备管置于洁净的1.5 ml 离心管(试剂盒内提供)中,在制备膜中央加25-30 μl Eluent 或去离子水,室温静置1 min。12,000×g 离心1 min 洗脱DNA。
* 将Eluent 或去离子水加热至65℃将提高洗脱效率。
Part IIIB. TA连接
【材料与方法】
pGEM-T Easy ligation kit(promega)
【步骤与方法】
1. 通过测量PCR产物A260nm计算PCR产物浓度
2. pGEM®-T 和pGEM®-T Easy 载体大概3kb 大小,系统提供的载体浓度为50ng/μl。计算连接反应中需要的PCR 产物的量采用以下公式:

根据推荐的插入片段:载体比例(3:1)加入足够的pGEM®-T 和pGEM®-T Easy载体进行连接,并进行对照反应。
3. 建立如下反应体系。注意使用低DNA 结合力的0.5ml 离心管(如VWR 目录号20170-310),每次使用2×快速连接缓冲液时要充分混匀。

4. 用移液器吹打连接反应使之混匀后,为得到最大数目的转化菌落,4℃孵育过夜。
Part ⅢC. 转化
【材料与试剂】
l Cells JM109 Takara 100μL×10tubes,10μL control vector(pUC19,0.1ng/μg)×1 tube,于﹣80℃。
l 每个连接反应准备1个LB/氨苄/IPTG/X-Gal平板,涂板前将平板平衡至室。(步骤 )
【步骤与方法】
1. 取6支1.5毫升离心管置于冰上分别标记“positive control”(此次实验为T质粒自连载体转化),“negative control”(此次实验为无插入片段质粒载体转化), “EGFP1”,“EGFP2”,“EGFP3”,“EGFP4”。
2. 将3管冻存的JM109高效率感受态细胞从﹣70℃冰箱中取出,放置在冰浴直至融化(大概5分钟),轻轻震荡离心管使之混匀。
3. 向步骤2准备的每个转化管中加入50μL感受态细胞。
4. 往实验组离心管中加入对应的5μL转化子。
5. 冰浴30 min。
6. 在精确的42℃水浴中热激60 sec(不要振动),后迅速置于冰上,冷却5 min。
7. 每管连接反应转化细胞中加入平衡至室温的0.5 ml SOC 培养基
8. 在37℃震荡培养60 min。
9. 将各管培养液均匀涂布在对应标号的LB/氨苄/IPTG/X-Gal平板中,将培养基倒扣于恒温箱中孵育12-16 hours。
Part ⅢD. 筛选
【材料与试剂】
TAKARA Taq system(order code R001A):Taq buffer(10×);dNTP(每种 2.5mM, 总共10mM,于﹣20℃下保存)。
【步骤与方法】
1. 如下建立预PCR混合液:
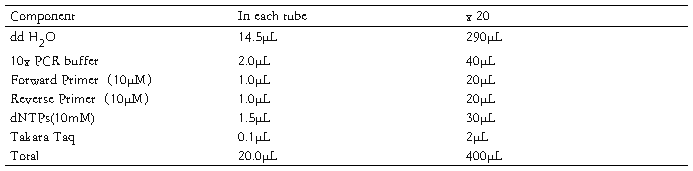
2. 将预PCR混合液每管20μL分装8管。
3. 用经灭菌的10μL枪头在EGFP系列培养基中分别挑取3个白色聚落和1个蓝色菌落,在positive control培养基中挑取一个白色菌落,在negative control培养基中挑取一个蓝色菌落,迅速地将其浸入对应编号的预PCR混合液中。
4. 进行PCR反应,参数如下:
预变性:95℃ 5 min
35个循环(变性;引物退火;引物延申):94℃ 30 sec;55℃ 30 sec;72℃ Amplicon specific(~1 minute per kb)
最后延伸:72℃ 5min。
5. 对PCR产物进行凝胶电泳分析。
Part ⅢD. 质粒提取
【材料与试剂】
l LB 培养基
l 转化菌落培养基
l TianGen Plasmid Mini kit
l ddH2O
【方法与步骤】
1. 柱平衡步骤:向吸附柱CP3 中(吸附柱放入收集管中)加入500μl 的平衡液BL,12,000 rpm (~13,400×g ) 离心1 分钟,倒掉收集管中的废液,将吸附柱重新放回收集管中。(请使用当天处理过的柱子)
2. 取1-5 ml 过夜培养的菌液,加入离心管中,使用常规台式离心机,12,000 rpm(~13,400×g ) 离心1 分钟,尽量吸除上清(菌液较多时可以通过多次离心将菌体沉淀收集到一个离心管中)。
3. 向留有菌体沉淀的离心管中加入250 μl 溶液P1(请先检查是否已加入RNaseA),使用移液器或涡旋振荡器彻底悬浮细菌沉淀。注意:如果有未彻底混匀的菌块,会影响裂解,导致提取量和纯度偏低。
4. 向离心管中加入250 μl 溶液P2,温和地上下翻转6-8 次使菌体充分裂解。注意:温和地混合,不要剧烈震荡,以免打断基因组DNA,造成提取的质粒中混有基因组DNA 片断。此时菌液应变得清亮粘稠,所用时间不应超过5 分钟,以免质粒受到破坏。如果未变得清亮,可能由于菌体过多,裂解不彻底,应减少菌体量。
5. 向离心管中加入350 μl 溶液P3,立即温和地上下翻转6–8 次,充分混匀,此时将出现白色絮状沉淀。12,000 rpm (~13,400×g )离心10 分钟,此时在离心管底部形成沉淀。注意:P3 加入后应立即混合,避免产生局部沉淀。如果上清中还有微小白色沉淀,可再次离心后取上清。
6. 将上一步收集的上清液用移液器转移到吸附柱CP3 中 (吸附柱放入收集管中),注意尽量不要吸出沉淀。12,000 rpm (~13,400×g)离心30-60 秒,倒掉收集管中的废液,将吸附柱CP3 放入收集管中。
7. 可选步骤:向吸附柱CP3 中加入500 μl 去蛋白液PD, 12,000 rpm(~13,400×g )离心30–60 秒,倒掉收集管中的废液,将吸附柱CP3 重新放回收集管中。如果宿主菌是end A+宿主菌(TG1,BL21, HB101,JM 系列, ET12567等),这些宿主菌含有大量的核酸酶,易降解质粒DNA,推荐采用此步。如果宿主菌是endA-宿主菌(DH5α,TOP10 等),这步可省略。
8. 向吸附柱CP3 中加入600 μl 漂洗液PW(请先检查是否已加入无水乙醇),12,000rpm (~13,400×g )离心30-60 秒,倒掉收集管中的废液,将吸附柱CP3 放入收集管中。
9. 向吸附柱CP3 中加入600 μl 漂洗液PW,12,000 rpm (~13,400×g) 离心30-60 秒,倒掉收集管中的废液。
10. 将吸附柱CP3 放入收集管中, 12,000 rpm (~13,400×g) 离心2 分钟,目的是将吸附
柱中残余的漂洗液去除。
注意:漂洗液中乙醇的残留会影响后续的酶反应(酶切、PCR 等)实验。为确保下
游实验不受残留乙醇的影响,建议将吸附柱CP3 开盖,置于室温放置数分钟,以彻
底晾干吸附材料中残余的漂洗液。
11. 将吸附柱CP3 置于一个干净的离心管中,向吸附膜的中间部位滴加50-100 μl 洗脱缓冲液EB,室温放置2 分钟,12,000 rpm (~13,400譯) 离心1 分钟将质粒溶液收
集到离心管中。
注意:洗脱缓冲液体积不应少于50 μl,体积过小影响回收效率。洗脱液的pH 值对
于洗脱效率有很大影响。若后续做测序,需使用去离子水做洗脱液,并保证其pH
值在7.0-8.5 范围内(可以用NaOH 将水的pH 值调到此范围),pH 值低于7.0 会降
低洗脱效率。且DNA 产物应保存在-20℃,以防DNA 降解。__
12. DNA产物进行凝胶电泳分析(PCR反应体系及参数参考Part ⅢD 步骤1.)
实验结果分析
参考文献