XXXXXXXXXXXXX公司
特殊过程确认报告
目 录
摘要............................................................................................................................................................1 1.
2.
3.
4.
5.
6.
7.
8.
目的..............................................................................................................................................1 介绍..............................................................................................................................................1 程序..............................................................................................................................................1 结果..............................................................................................................................................2 确认意见......................................................................................................................................4 确认的保持..................................................................................................................................4 再确认..........................................................................................................................................5 附录清单......................................................................................................................................5
摘要
XX于20xx年初次建立灭菌剂量,由于近几年产品不断增加、ISO标准换版以及辐照机构变更,20xx年XX按照ISO11137-2006的要求重新进行了灭菌确认。确认的内容包括:辐照机构、辐照剂量及辐照加工。确认结果表明:XX灭菌过程符合ISO标准要求,以后应按照规定的周期进行生物负载监测和剂量审核,当产品族、辐照条件发生重大变化时,应再确认。
1. 目的
XX在20xx年委托XX建立灭菌剂量,由于近几年XX产品的不断增加、国际标准及中华人民共和国药典的换版、辐照机构变更,XX在20xx年重新进行了灭菌确认,以证实产品辐照符合ISO11137-2006的要求,灭菌后的产品能达到10-6的无菌保证水平。
2. 介绍
本报告是根据ISO 11137-1:2006 保健产品的灭菌-辐射-第1部分:医疗器械灭菌过程的发展、验证和常规控制要求进行。为了完成灭菌确认,需完成以下分项:
? 辐照机构鉴定,由于XX委托专业的辐照机构完成灭菌,因此对其辐照资质、辐照加工
能力、辐照设备是否进行了安装鉴定、操作鉴定、性能鉴定予以确认,确认后委托其灭菌服务。
? 辐照剂量确定,产品灭菌前,应规定产品的最大可接受剂量、建立灭菌剂量。如果采用
25KGy 灭菌剂量辐照产品,应作证实。
? 辐照加工确定,确定装箱模式,进行剂量分布实验,按照要求完成产品的常规辐照。
3. 程序
3.1 辐照机构鉴定
辐照机构应具有合法有效的营业执照、射线安全许可证、质量体系证书,按照ISO11137的要求对辐照工程进行了安装鉴定、操作鉴定、性能鉴定,严格控制加工过程,有完善的产品合格放行管理文件、库房管理文件,有产品辐照技术指导文件,能提供剂量检测报告。
3.2 辐照剂量
应规定产品的最大可接受剂量,建立灭菌剂量(达到无菌水平的最小吸收剂量)。
3.2.1 最大可接受剂量
可以根据产品的特点、行业标准规定制定产品的最大可接受剂量,在产品的寿命期内,产
品应满足规定的使用功能。
3.2.2 建立灭菌剂量
按照ISO11137-2:2006的规定建立灭菌剂量应进行定义划分产品族并选择代表产品、选择实验方法、实验实施几个程序。
3.2.2.1 定义划分产品族并选择代表产品
根据产品原料的性质和来源、产品的构成、产品的设计和尺寸、生产工艺对所有的无菌包装产品进行定义划分。对各产品族产品进行分析,选择能够代表本族产品的产品,依照中华人民共和国药典2005版,附录XI J微生物限度检查法:细菌和ISO11737-1:2006,进行初始污染菌检测,实验得到各族代表产品的平均生物负载,选择最高生物负载的产品作为建立灭菌剂量的代表产品,即实验产品。
3.2.2.2 选择实验方法
建立灭菌剂量的实验方法有:利用生物负载数量或抗性信息建立剂量,或者选择15或25Kgy的灭菌剂量并证实,证实方法可采用方法1 、方法2、VDMAX25或VDMAX15。
3.2.2.3 实验实施
采用VDMAX25方法按照ISO11137-2:2006的规定进行实验,完成生物负载实验、生物负载回收率实验、剂量实验、无菌实验及无菌实验的验证实验。当实验结果被接受,则建立的灭菌剂量能够使产品的无菌保证水平达到10-6。生物负载实验依照中华人民共和国药典2005版和ISO11737-1:2006给出产品中自然存在的活微生物数,使用生物负载回收率调整过的最高批平均生物负载确定验证剂量,用验证剂量辐照产品,并做无菌实验和无菌实验的验证实验。
3.3 辐照加工确定
产品应按照规定的装箱模式进行装箱,并在确定的剂量场下进行产品辐照,辐照结果应满足辐照要求。
3.3.1 根据辐照技术条件要求和产品特点设计包装箱,规定装箱模式,并按照规定装箱。
3.3.2 确定辐照装置内产品在一定的装箱模式、辐照条件下的剂量场分布情况,进行剂量分布实验。将剂量计按三维空间以网格式布放,充分遍及整个辐照容器,剂量计的数量按照辐照容器的大小和辐照设备的设计,给出最大剂量、最小剂量及对应位置。
3.3.3 在确定好的装箱模式、辐照条件下辐照产品,辐照剂量符合设定的剂量范围。
4. 结果
4.1 辐照机构鉴定
辐照机构为XX,具有合法有效的营业执照、射线安全许可证、质量体系证书。
XX辐照灭菌采用的1#装置经过了安装鉴定、操作鉴定和性能鉴定,有完善的产品合格放行管理文件、库房管理文件,有产品辐照技术指导文件,能提供剂量检测报告。
辐照机构鉴定记录见附录A。
鉴定结论:经鉴定,XX公司满足XX灭菌需求,可以委托其进行灭菌加工。
4.2 辐照剂量确定
4.2.1 最大可接受剂量,依据XX规定:XX产品的最大可接受剂量为X KGy。
4.2.2 建立灭菌剂量
4.2.2.1 定义划分产品族并选择代表产品
根据产品的外形结构将XX产品分为三类:XX类、XX类、XX类,根据产品的材料、表面形态、产品构成、生产过程等因素将三类产品进一步划分为X个产品族,在各产品族中选取生物负载风险最大的X种产品进行了初始污染值检测,从中选择生物负载最高的产品,作为建立和审核灭菌剂量的代表产品。初始污染值检测结果表明,XX的生物负载值最高,平均Xcfu/件,因此选择XX为XX事业部建立和审核灭菌剂量的代表产品。
4.2.2.2 选择实验方法
根据建立灭菌剂量方法不同的流程、样品要求、剂量差异以及无菌包装产品现行辐照灭菌剂量为25Kgy的现状,选择了VDMAX25作为建立灭菌剂量的方法。
4.2.2.3 实验实施
选取3批XX产品(批号:XXXXX)共30件做生物负载实验,得到批平均生物负载分别为XXX(CFU/产品单元),生物负载回收率实验结果为XX,用回收率调整后的最高批平均生物负载值为XXCFU/产品单元,按照ISO11137-2:2006条款9计算无菌保证水平为10-1的验证剂量为XXKGy, 用验证剂量辐照XX批XX产品X件,辐照后做无菌实验,无菌实验依照中华人民共和国药典2005版无菌检查法:薄膜过滤法实施,并按照该方法规定的验证方法对同批X件产品进行了抑菌实验,实验结果为无菌实验没有阳性、阴性对照为阴性、阳性对照为阳性、不存在抑菌成分。产品辐照灭菌验证成功且符合ISO11137-2:2006的方法VDMAX25。 辐照剂量确定记录见附录B。
确认结论:辐照剂量的确定程序符合ISO11137:2006的要求,经确定的剂量可以用于产品的大规模辐照。
4.3 辐照加工确定
4.3.1 编写了装箱模式工艺文件,工艺文件按照XXX辐照装置尺寸、钴源情况、运行方式,结
合XX产品包装盒尺寸、材质、重量,制定了装箱模式要求。
4.3.2 按照装箱规定进行装箱后,在XX进行了剂量分布实验。实验选取X大箱产品上下码放,
再将整个产品空间三维分割:X轴5等分、Y轴3等分、Z轴6等分,得到168个交点,将剂量计布放在各交点,然后码放在一个辐照容器内以单一路径动态步进方式进行辐照,辐照后检测剂量计得到各点的吸收剂量 。
实验结果:最大吸收剂量XKGy,对应位置X。
最小吸收剂量XKGy,对应位置X。
不均匀度:X
辐照加工确定记录见附录C。
确认结论:辐照加工确定符合ISO11137:2006的要求,经确定的辐照加工方式可用于产品的日常辐照。
5. 确认意见
辐照机构、辐照剂量、辐照加工确定程序符合ISO11137:2006的要求,通过确认,并可以进行产品大规模日常辐照。
6. 确认的保持
通过生物负载监测、剂量审核及产品族的保持来确定灭菌剂量的持续有效,通过辐照条件的保持来确认辐照加工的持续有效。
6.1生物负载监测
建立灭菌剂量后每3个月抽取代表产品进行生物负载监测,实验依照中华人民共和国药典2005版附录XI J微生物限度检查法:细菌和ISO11737-1:2006进行,批平均生物负载应小于当初建立灭菌剂量时的生物负载值。
6.2剂量审核
建立灭菌剂量后每3个月抽取代表产品进行剂量审核,剂量审核按照ISO11137-2:2006要求需完成生物负载实验、剂量实验及无菌实验。剂量审核成功灭菌剂量持续有效。
6.3 产品族的保持
每年对产品族进行复查,以确保产品族和代表产品族的产品持续有效。
生产过程中,对于生产条件、生产工艺发生改变的产品及新产品进行初始污染检测,确保
代表产品的持续有效。
6.4辐照条件的保持
检查辐照条件,当辐照条件发生变化时,根据影响的结果进行剂量分布实验或再确认或更换辐照机构。
7. 再确认
当辐照机构发生变化时,需进行辐照机构确认、辐照加工确认。
当代表产品发生变化、剂量审核失败导致重新建立灭菌剂量时,需进行辐照剂量确认。 当辐照条件发生变化时,需进行辐照加工确认。
8. 附录清单
附录A: 辐照灭菌确认——辐照机构鉴定
1.
2.
3.
4.
5.
6. 企业法人营业执照 辐射安全许可证 质量体系证书 Υ辐照装置验收纪要 辐照装置辐照加工过程确认报告 说明
附录 B: 辐照灭菌确认——辐照剂量确定
1.
2.
3.
4. 辐照灭菌确认——定义产品族与选择代表产品 辐照灭菌确认——选择建立灭菌剂量的方法 实验室认可证书 建立灭菌剂量实验合同
5. 建立灭菌剂量实验报告
附录C: 辐照灭菌确认——辐照加工确定
1. 灭菌产品装箱工艺规程
2. 剂量分布实验报告
第二篇:植入医疗器械辐照灭菌确认报告
辐照灭菌 确 认 报 告
拟 制 ****** 日 期 20xx年9月20日审 核 日 期 20xx年9月20日 批 准 ****** 日 期 20xx年9月20日 版 号 生效日期 20xx年10月1日
*******有限公司
1.主要内容和适用范围
本文规定了灭菌的验证和日常管理。
1.1 验证组成人员
2.辐照灭菌剂量的设定
验证的原理是基于ISO11137方法,即先对辐照前产品的初始污染菌进行测定,然后选择验证剂量。再用验证剂量对产品进行辐照,并测定存活微生物的样品件数,以此来确定最低灭菌剂量(SAL=10-6)。
2.1 方法
收集常规生产的标准包装产品,于灭菌前对三个批号进行随机抽样。其中取样比例(SIP)为1。
2.1.1初始污染菌的测定
根据每个样品的测试结果,计算出每件产品的平均带菌数。同时进行初始污染菌回收率的测试,以对该产品带菌数测试方法的有效性和重复性进行确认。初始污染菌的菌数取自三个独立批的单位产品的总平均带菌数。
试验方法:(平板计数法)
1. 洗脱液:
2. 样品处理:。
3. 需气菌培养:
4. 霉菌培养:
5. 计数:
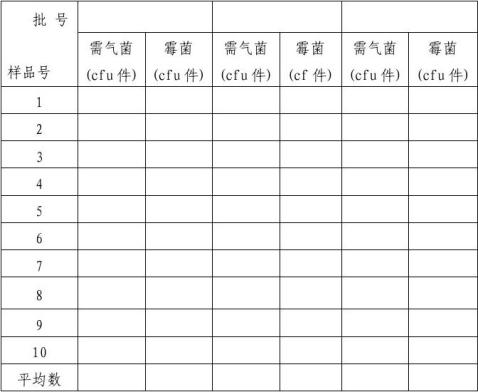
结果:参见下表:
阴性对照<10cfu/样品
三批产品初始污染菌测试结果如下:
批 号 初始污染菌平均数(cfu/件) 2.12验证剂量的选定
根据IOS11137:2006方法1中的表5,选择合适的验证剂量。医疗保健产品辐照灭菌的有效性确认和常规控制的要求(ISO11137)规定,应用校正后的总平均带菌数确定验证剂量,除非,某一批号的平均数的平均带菌数大于等于校正后的总平均带菌数的二倍。在此情况下,采用平均数最大批的数据确定验证剂量。
初始污染菌:
经三批连续产品初始污染菌及回收率的检测,每批产品初始污染菌如
下:
三批产品初始污染菌测试结果如下:
批号 初始污染菌平均数(cfu/件)
校正后平均初始污染菌数: 75
按照ISO11137 方法1 查得校正后总平均带菌数75cfu/件的验证剂量为7.6kGy。再按要求对100件产品采用7.6 kGy±10%的验证剂量进行辐射处理,再经无菌检查。根据以上说明,验证剂量为7.6kGy±10%,对100件产品进行辐照,剂量为6.84~8.36kGy。
从一个单独批号采样100件产品,在上海核新辐射厂进行辐照。所照剂量用该公司放置的剂量计测定,保证所测剂量落在规定剂量的±10%之内。
2.13 产品释出物的检验
方法:
1.标准菌株准备:取枯草杆菌黑色变种(ATCC9372),白色念珠菌(ATCC10231),传代培养,制成100cfu/ml备用。
2.产品处理:取灭菌产品以无菌操作转移到100mlSCDB培养基中,30~35℃培养24h。
3.接种:以无菌操作接种标准菌于上述产品SCDB培养基中,28~32℃培养出现阳性结果或是至少培养7天。用标准平板计数法进行微生物计数(CFU)。
结果:
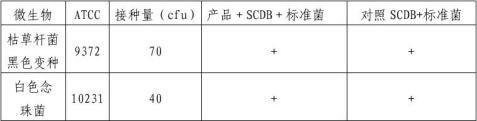
结论:经检测,该产品中未检出影响微生物生长的释出物。通过的细菌/真菌试验发现无抑制作用。
2.14 产品无菌检测
试验方法:
1.样品测试接种均应按无菌操作法(在100级净化工作台内)进行。
2.无菌打开包装,用灭菌剪刀、镊子,将产品转移至SCDB培养基中。
3.将样品培养基放28-32℃温箱中培养14天。
4.观察有无细菌生长。
结果:经试验结果如下表
无菌试验检验结果

结论:经测试,试验产品为无菌产品。既100件样品经验证剂量辐照后的无菌检查均为阴性。
2.15结论
剂量验证实验结果符合ISO11137中规定的合格标准。这表明,在本研究条件下,20.7kGy可确定为常规灭菌的最低剂量,此剂量提供的灭菌保证水平为SAL=10-6。
3 辐射灭菌加工确认
灭菌剂量20.7~40.0kGy的条件下,通过对该产品辐射灭菌过程的有效性进行确认,确定辐射灭菌过程运行参数,保证产品的灭菌质量。
3.1辐照试验装置的概况
1号装置设计装源量100万居里(37PBq),现实际装源量为66.05万居里(24.44PBq)(2006.6.21)。装置设计为6通道48工位自动化运行辐照装置,可实现自动换面,辐照吊箱尺寸为2250×760×560mm,辐射源为单板框架,板架尺寸为2800×1900mm,由上、中、下三排源架组成,每排设80个源孔,共计240孔,通过卷扬机实现辐射源的升降,提升速度为1.1
米/秒,目前总活度66.05万居里(24.44PBq)(2006.6.21),占用143个源孔,源到位重复性0.3%。
3.2 测量仪器及试剂
仪器:紫外分光光度计(UV-2401,每年定期校验)
剂量计:重铬酸钾(银)不确定度为±5%,每年定期与国家计量院进行对比,且批与批之间也进行比对。
3.3 确认设计
产品箱尺寸,产品密度。灭菌负载为******(******膏):
灭菌包装形式:
内包装:软管。
小包装材料:纸盒,每盒1支。
外包装材料:双瓦楞纸板,每箱100支。
产品装载模式及剂量计放置
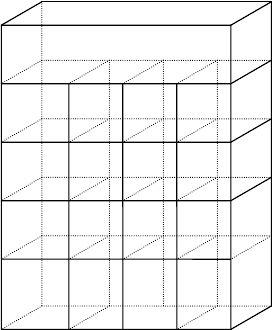
图一、产品装载模式
图二、剂量计放置
参照安装鉴定吊箱中的剂量分布,将该产品在1号装置运行参数确定每工位停留时间为20分00秒。
3.4 实验数据
产品在吊箱中吸收剂量的分布(1)
剂量计名称: 测量仪器:
剂量计批号: 测量温度:
吊箱 尺寸: 产品名称:
产品箱尺寸: 装载模式:
产品 密度: 运行模式:
测量结果


注:1.No.表示剂量计编号,D表示剂量计吸收剂量。
2.源位高度为7.14mm
3.常规监测点剂量计剂量吸收量Ds=32.34kGy
4.源的活度为59.64万Ci(22.07PBq)
结论:辐照吊箱中最大吸收剂量Dmax=30.65kGy,最小吸收剂量Dmin=21.74 kGy,不均匀U=Dmax/Dmin=1.410;最小吸收剂量与常规监测点处的吸收剂量之间的关系r=Dmin/Ds=0.672,R=Dmax/ Ds=0.948.
产品在吊箱中吸收剂量的分布(2)
剂量计名称:重铬酸钾(银) 测量仪器:UV-2401 剂量计批号:070104 测量温度:25℃
吊箱 尺寸:2350×760×560mm 产品名称:****** 产品箱尺寸:440×460×170mm 装载模式:16箱
产品 密度:0.116g/cm3 运行模式:6路20分00秒 测量结果
注:1.No.表示剂量计编号,D表示剂量计吸收剂量。
2.源位高度为7.14mm
3.常规监测点剂量计剂量吸收量Ds=32.44kGy
4.源的活度为59.64万Ci(22.07PBq)
结论:辐照吊箱中最大吸收剂量Dmax=30.17kGy,最小吸收剂量Dmin=21.86 kGy,不均匀U=Dmax/Dmin=1.380;最小吸收剂量与常规监测点处的吸收剂量之间的关系r=Dmin/Ds=0.674,R=Dmax/ Ds=0.930.
3.5 结论
根据上述产品中吸收剂量分布的测量结果,采用该设计条件能满足对
该产品的灭菌要求。因放射源的活度随时间的延长不断衰减,必须定期对放射源的活度进行校准,根据60Co γ放射源的半衰期为5.27年推算,每月的校准系数约为1.05%。
对于新产品或包装、装载形式、设备和过程参数发生改变时,应重新验证,除非经过论证与以前验证过的产品、包装形式或装载形式具有等效性。
4 日常控制
应制定灭菌过程日常工艺操作的书面程序。操作人员应严格执行按工艺操作规程执行,应作记录。以证明其在规定工艺范围内运行。
每次灭菌后,应抽取规定数量的产品,对产品做无菌的出厂检验,并出具报告。
5产品放行
6 验证日期:
7 验证单位:
8 ******灭菌工艺流程图