植化方法学实验报告
单位:六班七组
成员:樊若西 付云飞 原雪娜 邹晓楠 毕天民
总结汇报:毕天民
报告执笔:付云飞
20##年7月8日
大黄中蒽醌类成分的提取、分离和鉴定
一、实验题目:大黄中蒽醌类成分的提取、分离和鉴定
二、实验目的:
1、 掌握大黄浸膏的提取方法
2、 了解柱层析、薄层层析的操作技术
3、 学习用硅胶柱色谱法和硅胶制备薄层色谱法分离大黄中的蒽醌类成分的提取分离方法
4、 学习蒽醌类化合物的鉴定方法
三、实验原理:
大黄记载于《神农本草经》等许多文献中,用于泻下、健胃、清热、解毒等。自古以来,大黄在植物性泻下药中占有重要位置,是一位很早就被各国药店所收载的世界性生药。大黄的种类繁多,优质大黄是蓼科植物掌叶大黄(Rheumpalmatclm L),大黄(R.officinale Baill)及唐古特大黄(R.tangutium Maxim.et Regll)的根茎及根大黄中含有多种游离的羟基蒽醌类化合物以及它们与糖所形成的苷,总含量约2-5%。已经知道的羟基蒽醌主要有下列五种:
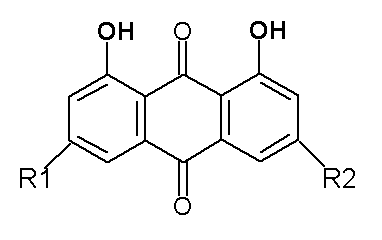
大黄酸 R1=H R2=COOH
大黄素 R1=CH3 R2=OH
芦荟大黄素 R1=CH2OH R2=H
大黄素甲醚 R1=CH3 R2=OCH3
大黄酚 R1=CH3 R2=H
大黄中蒽醌苷元,其结构不同,因而酸性强弱也不同。大黄酸连有-COOH,酸性最强;大黄素连有β-OH,酸性第二;芦荟大黄素连有苄醇-OH,酸性第三;大黄素甲醚和大黄酚均具有1,8-二酚羟基,前者连有-OCH3和-CH3,后者只连有-CH3,因而后者酸性排在第四位。
四、实验材料:
1、 仪器:旋转蒸发仪、红外灯、天平、回流装置一套、圆底烧瓶、烧杯、蒸发皿、层析槽、布氏漏斗、抽滤瓶、试管、滴管、橡皮管、分液漏斗、普通滤纸、水浴锅、薄层板、喷雾器、点样毛细管等。
2、 试药:大黄
3、 试剂:95%乙醇、石油醚、氯仿、乙酸乙酯、丙酮、甲醇、冰醋酸
4、 吸附剂:薄层色谱硅胶(10~40目)
柱色谱硅胶(200~300目)
显色剂(装置):1%氢氧化钾溶液,2%三氯化铁溶液,10%硫酸乙醇溶液,紫外灯
五、实验步骤:
(一)大黄浸膏的提取
称取大黄50g,用95%乙醇200ml加热回流提取2h,常压蒸馏回收溶剂,得大黄浸膏47.6g。
(二)硅胶柱色谱
1、 装柱:取一玻璃层析柱,垂直固定在铁架台上,在管的下端垫入少量棉花,装入200~300目的硅胶45g,湿法装柱,层析柱柱长约50cm,内径3cm。
2、 拌样:取大黄浸膏1.5g置于小蒸发皿中,加少量甲醇(刚溶为度),用滴管加到4.5g的100~140目硅胶上,仔细拌匀,挥干,碾细。
3、 上样:湿法上柱,检漏后,小心将拌样硅胶装入柱。
4、 洗脱:将色谱柱活塞打开,洗脱剂为石油醚-乙酸乙酯混合液
5、 收集:各流份接入试管,洗脱液分别收集,合瓶水浴浓缩,分别通过TLC检查,与标准品对照,合并相同组分,分别得到粗品。
吸附剂:硅胶G
展开剂:石油醚-乙酸乙酯-冰醋酸
A、以石油醚:乙酸乙酯=50:1(滴入2滴冰醋酸)为流动相洗脱,点板,无荧光反应;
B、以石油醚:乙酸乙酯=30:1(滴入2滴冰醋酸)为流动相洗脱,点板,有荧光反应,点板合瓶,旋蒸转移到小瓶中,制备薄层分离两个成分;
C、以石油醚:乙酸乙酯=15:1(滴入2滴冰醋酸)为流动相洗脱,点板,无荧光反应;
D、以石油醚:乙酸乙酯=10:1(滴入2滴冰醋酸)为流动相洗脱,点板,无荧光反应;
E、以石油醚:乙酸乙酯=5:1(滴入2滴冰醋酸)为流动相洗脱,点板,有荧光反应,点板合瓶,旋蒸转移到小瓶中,制备薄层分离两个成分;
F、以石油醚:乙酸乙酯=2:1(滴入2滴冰醋酸)为流动相洗脱,点板,无荧光反应;
G、以石油醚:乙酸乙酯=1:1(滴入2滴冰醋酸)为流动相洗脱,点板,无荧光反应。
(三)薄层色谱
A、以石油醚:乙酸乙酯=10:1(加2滴冰醋酸)为展开剂,样品点样,分别刮板,碾碎硅胶,溶于甲醇,过滤,挥干,
B、以石油醚:乙酸乙酯=2:1(加2滴冰醋酸)为展开剂,样品点样,分别刮板,碾碎硅胶,溶于甲醇,过滤,挥干,
C、以石油醚:乙酸乙酯=2:1(加2滴冰醋酸)为展开剂,样品点样,分别刮板,碾碎硅胶,溶于甲醇,过滤,挥干,
(四)与标准品对照
纯化后各成分同对照品点板,石油醚:乙酸乙酯=2:1(加2滴冰醋酸)展开,荧光点单一、对应。荧光颜色?
(五)样品处理
分别刮板,碾碎硅胶,甲醇溶解,充分沉淀,过滤,挥干后,溶于色谱甲醇,充分溶解,过0.45微孔滤膜,
(六)HPLC鉴定
流动相条件:YMC-C18柱,流速1.0ml/min,检测波长254nm
流动相:90%甲醇:水:磷酸=85:15:0.1
进样量:20µl
冲柱子:开机,甲醇冲10min,流动相跑基线平衡;10%甲醇冲30min,甲醇冲30min,关机。
六、结果与讨论:
1、溶剂系统的选择
在高速逆流色谱中 ,溶剂系统的选择是相当重要的步骤 ,它直接影响出峰顺序与时间。但是由于本实验所要分离的五种成分极性相差较大,仅仅通过调节溶剂组成的方法并不能达到理想的分离效果。加入酸碱调节后 ,加大了上下相极性的差别 ,可以得到所要分离的三种成分 (大黄酸、 大黄素和芦荟大黄素 );大黄素甲醚和大黄酚也得到了很好的分离 ,只是用下相很难洗脱出。当前三个组分流出后 ,换用上相做流动相 ,利用后两种极性较弱 ,采用反转的方法可以得到很好的分离峰.
2、成分鉴定
经过与标准品的对照,证实了分离出的四个成分成分单一,分别为大黄素、大黄酚、大黄酸、芦荟大黄素;大黄粗提物中所分离出的四个成分含量分别为:大黄素91.9%,大黄酚99.1%,大黄酸98.6%,芦荟大黄素92.3%。
七、结论:
利用加入酸碱调节的冰醋酸分离大黄的蒽醌类物质 ,效果是相当理想的 ,分离纯化过程可以一次完成 ,得到大黄的五种活性成分纯品。而采用传统方法结合柱色谱需要多次重结晶纯化 ,才能得到 ,这表明高速逆流色谱为天然产物的分离制备开辟了一个新途径。
八、注意事项:
1、填充操作的要求:填料颗粒不破碎,但又填得均匀,松紧适度,湿法装柱要尽量赶尽气泡,不留任何间隙。还要注意柱头抵柱物(玻璃棉的洁净,用量宜越少越好)。
2、上柱时要多留一段液体,拌样硅胶加入后还有一定的距离(有颜色应放掉,直至无色),盖样硅胶扩大原点。
4、由于色谱柱属于吸附性物质,柱子上时间长,扩散长,比例前紧后松(极性大的比例大则洗脱不好),所以长时间不用要通一段时间气进行老化后再使用,否则容易造成基线不稳或噪音大、灵敏度低等现象;如果色谱柱受到污染,它表现出的明显现象是峰分离变差,灵敏度变低并且峰形托尾,所以解决这种污染柱的根本方法是更换色谱柱。
2、混合溶剂洗脱,流速控制1mL/min,不能过快。流分每15mL收集一份,薄层检识后,相同流分再合并。
3、控制点样量,注意点样斑点位置;仔细观察斑点、溶剂前沿移动距离。
4、展开剂加入酸是为了防止拖尾。
5、有色带看分离,色带可能是色素杂质;荧光反应亮蓝色除非是香豆素,否则一般为杂质,不是成分。
6、处理样品时,甲醇不是最好的,粘合剂CMC-Na在甲醇中会溶解,有别的则不会选甲醇,丙酮好些,氯仿挥发太大。
7、制备薄层要预饱和,比较同一性要求做预饱和,否则分离不好拖尾。
8、析晶一般较纯,蒽醌苷元爱析出晶体,但有可能是两个物质的混晶,单体析晶纯度高。
9、硅胶ODS薄层板根本原理不同,所以分离不同。Rf值:大黄酚>大黄素甲醚>大黄素>芦荟大黄素>大黄酸;HPLC出峰顺序:芦荟大黄素>大黄酸>大黄素>大黄酚>大黄素甲醚,两个系统不仅仅是正反顺序对调,如果只是一正一反一点意义都没有,要换系统。
10、要注意实验的计划性(例如制备薄层的时候,如果没时间展开,但是又提前点板了,是会有吸附的,影响实验结果)。
第二篇:实验报告 蛋白质分子的测定
实验一 蛋白质分子的测定─凝胶层析法
一、 实验原理
凝胶层析法(即凝胶过滤法,gel filtration)是利用凝胶把分子大小不同的物质分离开的一种方法。将凝胶颗粒在适宜溶剂中浸泡,使充分吸液膨胀,然后装入层析柱中,加入欲分离的混合物,再以同一溶剂洗脱,在洗脱过程中大分子不能进入凝胶内部而沿凝胶颗粒间的空隙最先流出柱外,小分子可以进入凝胶内部,流苏缓慢,一直最后流出柱外,从而使样品中分子大小不同的物质得以分离。
凝胶是由胶体溶液凝结而成的固体物质,不论是天然凝胶还是人工合成凝胶,其内部都具有很微细的多空网状结构。凝胶层析法常用的天然凝胶是琼脂糖凝胶(Sepharose),人工合成的凝胶是聚丙烯酰胺凝胶(Bio-gel-P)和葡聚糖凝胶(Sephadex G)。其中葡聚糖凝胶是具有不同孔隙度的立体网状结构的凝胶,不溶于水。
将凝胶装柱后,柱床体积称为“总体积”,以Vt表示。Vt由Vo,Vi与Vg三部分组成,即Vt=Vi+Vg+Vo。Vo为“孔隙体积”、“外水体积”,即存在于柱床内凝胶颗粒外面空隙之间的水相体积;Vi为内体积,即凝胶颗粒内部所含水相的体积;Vg为凝胶本身体积;Ve为洗脱体积,即自加入样品时算起到组分最大浓度(峰)出现时所流出的体积,Ve与Vo及Vi之间的关系为:Ve=Vo+KdVi,;Kd为样品组分在二相间的分配系数,Kd=(Ve-Vo)/Vi,有效分配系数为Kav,Kav=(Ve-Vo)/(Vt-Vo)。
在一般情况下,凝胶对组分没有吸附作用时,当流动相流过Vt体积后,所有的组分都应该被洗出来,这一点为凝胶层析的特点,与一般层析方法不同。
Ve与分子量的关系:对同一类型的化合物,洗脱特性与组分的分子量有关,流过凝胶柱时,按分子量大小顺序流出,分子量大的走在前面。Ve与分子量的关系为:Ve=K1-K2logM,K1与K2为常数,M为分子量,通常用Kav代替Ve,建立标准蛋白质分子式量LgM与Kav的标准曲线,称为“选择曲线”。在允许的工作范围内,曲线越陡,则分级越好,而工作坊为越窄。凝佼层析主要决定于溶质分子的大小,每一类型的化合物,如球蛋白类,右旋糖酐类等都有它自己的特殊的选择曲线,可用以测定未知物的Mr,测定时以使用曲线的直线部分为宜。
二、 试剂材料
[试剂]
1、标准蛋白质:蓝色葡聚糖(分子式量200000),牛血清蛋白(分子式量67000),胃蛋白酶(分子式量36000),CytC(分子式量13700)。
2、洗脱液:0.2mol/L Tris-HCl-KCl,pH7.5
3、Sephadex G-75。
[器材]
1、层析柱:柱管1.0×40cm。
2、紫外分光光度计。
3、部分收集器。
4、刻度试管。
三、 实验步骤
[1] 凝胶装柱:先将洗脱液缓慢导入柱管内,然后将处理好的凝胶溶液加入层析管中。等到凝胶层胶面快接触到管顶端时,将洗脱管接好,洗脱30分钟,直到凝胶柱稳定。
[2] 调试液滴速度:将洗脱液速度调节至每分钟17滴,将时间定在3.0分钟自动调换试管,为加样收集洗脱液做准备。
[3] 加样:当液滴速度稳定在17滴每分钟之后,将层析管打开,用胶头滴管吸走多余的洗脱液。此时将蓝色葡聚糖沿管壁加入凝胶层,同时用夹子夹住洗脱液出口,当葡聚糖加完之后,松开夹子,记录蓝色葡聚糖达到层析管低端的时间,此时收集的洗脱液体积即为Vo,Vo=VeI。此时用夹子夹住洗脱液出口,重复上述步骤,添加牛血清蛋白质。按照上述方法将4种标准蛋白质样本依次加入层析管中层析。需要注意的是,需记录每次加样的试管号,以便计算各种物质的洗脱液体积,以后每加一种蛋白质的时间都要比上一种蛋白质添加时间间隔长;用分光光度计测定各试管洗脱液吸光度值,标出各个峰值,第二次加样到第二个峰值出现的各试管洗脱液的体积即为VeII,第三次加样到第三个峰值出现的各试管洗脱液的体积即为VeIII,第四次加样到第三个峰值出现的各试管洗脱液的体积即为VeIV。
[4]记录上述试验各部实验数据,建立有效分配系数Kav和洗脱液Ve标准曲线。
四、 实验结果
实验过程中,记录实验试管编号和每个试管的洗脱液体积,以及相对应的吸光度值如下表所示:
表1 凝胶层析实验原始数据
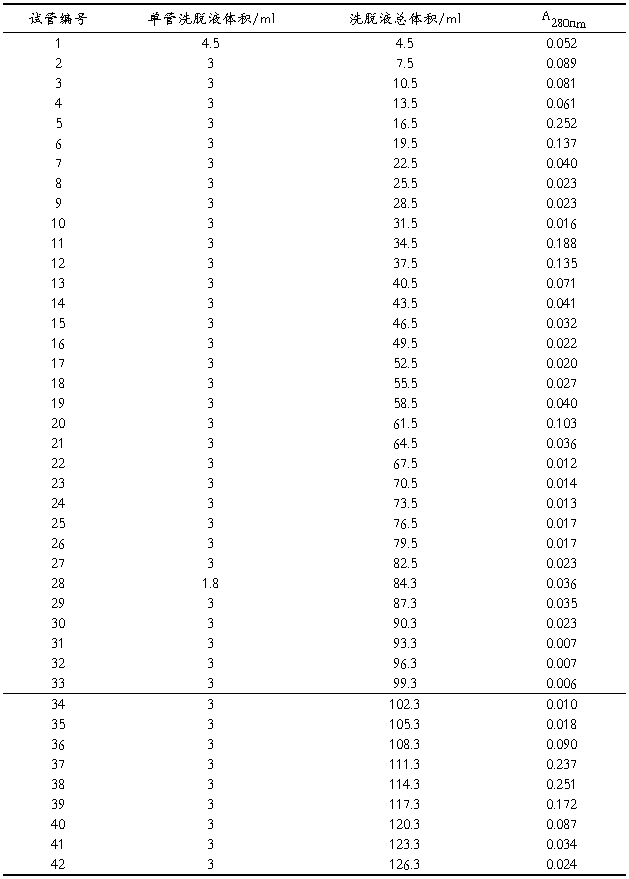
利用Excel2003绘制洗脱液总体积—吸光值散点图,见图1。和实验操作一样,有4个峰值,分别为蓝色葡聚糖、牛血清蛋白、胃蛋白和CytC。从中可知,V0=VeI=16.5ml,VeII=34.5ml,VeIII=61.5ml,VeIV=114.3ml。

图1 凝胶层析洗脱液体积与对应吸光度值散点图
建立标准蛋白Ve与与LogM标准曲线,见表2、图1。相关系数R2 = 0.9267,相关性较好,可作为为测定未知蛋白的标准曲线。因此最终的标准曲线为:Ve= -84.062·LogM+452.21。
表2 各标准蛋白Ve与logM值

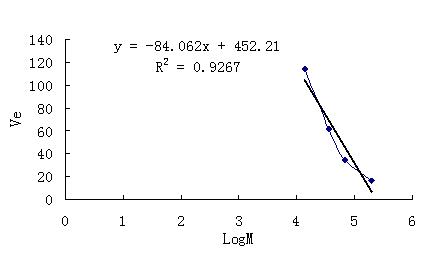
图2 蛋白质Ve与LogM的线性图
五、 分析与讨论
[1] 在允许的工作范围内,曲线越抖,则分级越好,本实验选择曲线斜率为-84.062,曲线很陡,说明此蛋白质的分级很好。
[2] 从洗脱液总体积—吸光值散点图(图1)看,除了4个标准品的峰外,在葡聚糖之前、胃蛋白和CytC之间均出现了很小的波峰,干扰实验结果分析。出现这种情况的原因可能是加样不均匀或者仪器本身不稳定等。
[3] 实验的误差原因及注意事项有:凝胶干粉要充分溶胀;层析柱粗细要均匀;连续用的小乳胶管不能有破损;实验过程中要保持层析柱液面水平;加样的浓度和粘度要适中;洗脱用的液体应与凝胶溶胀所用液体相同;收集的溶液体积测量要准确等。