第一章绪论
1.1 高分子的基本概念、特点
高分子化学:是研究聚合反应机理和动力学,聚合反应与聚合物的分子量和分子量分布,以及聚合物结构之间关系的一门学科。
单体:能通过相互反应生成高分子的化合物。
高分子或聚合物:由许多结构和组成相同的单元相互键连而成的相对分子质量在10000以上的化合物。相对分子质量低于1000的称为低分子。相对分子质量介于高分子和低分子之间的称为低聚物(又名齐聚物)。相对分子质量大于1 000 000的称为超高相对分子质量聚合物。
主链:构成高分子骨架结构,以化学键结合的原子集合。
侧链或侧基:连接在主链原子上的原子或原子集合,又称支链。支链可以较小,称为侧基;也可以较大,称为侧链。
聚合反应:由低分子单体合成聚合物的反应称做~.
重复单元:聚合物中组成和结构相同的最小单位称为~,又称为链节。
结构单元:构成高分子链并决定高分子性质的最小结构单位称为~
单体单元:聚合物中具有与单体的化学组成相同而键合的电子状态不同的单元称为~。
连锁聚合(Chain Polymerization):活性中心引发单体,迅速连锁增长的聚合。烯类单体的加聚反应大部分属于连锁聚合。连锁聚合需活性中心,根据活性中心的不同可分为自由基聚合、阳离子聚合和阴离子聚合。
逐步聚合(Step Polymerization):无活性中心,单体官能团之间相互反应而逐步增长。绝大多数缩聚反应都属于逐步聚合。
加聚反应(Addition Polymerization):即加成聚合反应, 烯类单体经加成而聚合起来的反应。加聚反应无副产物。
缩聚反应(Condensation Polymerization):即缩合聚合反应,单体经多次缩合而聚合成大分子的反应。该反应常伴随着小分子的生成。
聚合反应 (Polymerization):由低分子单体合成聚合物的反应。
线型聚合物:指许多重复单元在一个连续长度上连接而成的高分子.
热塑性塑料(Thermoplastics Plastics):是线型可支链型聚合物,受热即软化或熔融,冷却即固化定型,这一过程可反复进行。聚苯乙烯(PS)、聚氯乙烯(PVC)、聚乙烯(PE)等均属于此类。
热固性塑料(Thermosetting Plastics):在加工过程中形成交联结构,再加热也不软化和熔融。酚醛树脂、环氧树脂、脲醛树脂等均属于此类。
1.2 高分子化合物的分类
1) 按高分子主链结构分类:可分为:①碳链聚合物(Carbon-chain Polymer):大分子主链完全由碳原子组成的聚合物。②杂链聚合物(Hetero-chain Polymer):聚合物的大分子主链中除了碳原子外,还有氧、氮,硫等杂原子。③元素有机聚合物 (Element Organic Polymer) :聚合物的大分子主链中没有碳原子孙,主要由硅、硼、铝和氧、氮、硫、磷等原子组成。④无机高分子(Inorganic Polymer):主链与侧链均无碳原子的高分子。
2)按用途分可分为:塑料、橡胶、纤维三大类,如果再加上涂料、粘合剂和功能高分子则为六大类。塑料(Plastics):具有塑性行为的材料,所谓塑性是指受外力作用时,发生形变,外力取消后,仍能保持受力时的状态。塑料的弹性模量介于橡胶和纤维之间,受力能发生一定形变。软塑料接近橡胶,硬塑料接近纤维。 橡胶(Rubber):具有可逆形变的高弹性聚合物材料。在室温下富有弹性,在很小的外力作用下能产生较大形变,除去外力后能恢复原状。 橡胶属于完全无定型聚合物,它的玻璃化转变温度(T g)低, 分子量往往很大,大于几十万。 纤维(Fiber):聚合物经一定的机械加工(牵引、拉伸、定型等)后形成细而柔软的细丝,形成纤维。纤维具有弹性模量大,受力时形变小,强度高等特点, 有很高的结晶能力,分子量小,一般为几万。
3)按来源分可分为:天然高分子、合成高分子、半天然高分子(改性的天然高分子)
4)按分子的形状分:线形高分子、支化高分子、交联(或称网状)高分子
5)按单体分:均聚物、共聚物、高分子共混物(又称高分子合金)
6)按聚合反应类型分:缩聚物、加聚物
7)按热行为分:热塑性聚合物(Thermoplastics Polymer):聚合物大分子之间以物理力聚集而成,加热时可熔融,并能溶于适当溶剂中。热塑性聚合物受热时可塑化,冷却时则固化成型,并且可以如此反复进行。 热固性聚合物(Thermosetting Polymer):许多线性或支链形大分子由化学键连接而成的交联体形聚合物,许多大分子键合在一起,已无单个大分子可言。这类聚合物受热不软化,也不易被溶剂所溶胀。
8)按相对分子质量分:高聚物、低聚物、齐聚物、预聚物。
1.3 相对分子质量及其分布
1)相对分子质量
平均相对分子质量:相对于一般低分子化合物都具有确定的相对分子质量而言,一般合成聚合物都不是由具有相同相对分子质量的大分子组成,而是由许多相对分子质量大小不等的同系物分子组成的混合物。因此,高分子化合物的相对分子质量只是这些同系物相对分子质量的统计平均值。
数均分子量: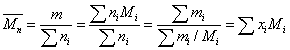
 为i-聚体的分子分率和质量分率。某体系的总质量m为分子总数所平均。.
为i-聚体的分子分率和质量分率。某体系的总质量m为分子总数所平均。.
质均分子量:采用光散射法测得: 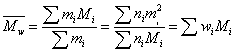
粘均分子量(Viscosity-average Molecular Weight):用粘度法测得的聚合物的分子量。 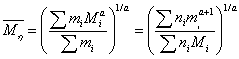
2)聚合度
聚合度( ):即高分子链中重复单元的重复次数,以
):即高分子链中重复单元的重复次数,以 表示;衡量聚合物分子大小的指标。
表示;衡量聚合物分子大小的指标。
聚合度与相对分子质量的关系为
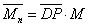 式中M为重复单元的相对分子质量.
式中M为重复单元的相对分子质量.
由于共聚物和混缩聚物的重复单元由两个或两个以上结构单元组成,如果采用聚合度往往会带来计算上的不便,因此大部分情况下,将聚合度定义为每个大分子链所含结构单元数目的平均值,通常以表示。
聚合度与相对分子质量的关系为
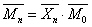 ,
,  为结构单元的平均相对分子质量。
为结构单元的平均相对分子质量。
特别强调:聚合度的计算最好以结构单元数目而不以重复单元数目为基准,即通常采用的是表示聚合度。
3) 相对分子质量分布
多分散性(Polydispersity):聚合物通常由一系列相对分子量不同的大分子同系物组成的混合物,这种相对分子质量的不均一性称为相对分子质量的多分散性。
多分散性有三种表示法:①多分散系数;②分级曲数;③分布函数。
多分散系(指)数可以用重均分子量和数均分子量的比值来表示,这一比值称为多分散指数,其符号为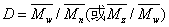 ,对于完全单分散的聚合物D=1,其数值大小表征聚合物相对分子质量大小悬殊的程度。
,对于完全单分散的聚合物D=1,其数值大小表征聚合物相对分子质量大小悬殊的程度。
分子量分布(Molecular Weight Distribution, MWD ):由于高聚物一般由不同分子量的同系物组成的混合物,因此它的分子量具有一定的分布,分子量分布一般有分布指数和分子量分布曲线两种表示方法。
第二章 逐步聚合
2.1 逐步聚合反应的基本概念
1 逐步的特征
逐步聚合(Step Polymerization):通常是由单体所带的两种不同的官能团之间发生化学反应而进行的。无活性中心,单体官能团之间相互反应而逐步增长。绝大多数缩聚反应都属于逐步聚合。
其特征为:①逐步聚合反应是通过单体功能基之间的反应逐步进行的。在反应初期,聚合物远未达到实用要求的高分子量(>5000——10000)时,单体就已经消失了。②逐步聚合反应的速率是不同大小分子间反应速率的总和。③聚合产物的相对分子质量随转化率增高而逐步增大的。④在高转化率才能生成高分子量的聚合物。
2逐步聚合反应的分类
1)按反应机理分类
逐步缩聚反应:带有两个或两个以上官能团的单体之间连续、重复进行的缩合反应,即缩掉小分子而进行的聚合。反应过程中,不小分子副产物生成。
逐步加成聚合:单体分子通过反复加成,使分子间形成共价键,逐步生成高相对分子质量聚合物的过程,其聚合物形成的同时没有小分子析出,如聚氨酯的合成。逐步聚合反应的所有中间产物分子两端都带有可以继续进行约定缩合反应的官能团,而且都是相对稳定的。当某种单体所含有官能团的物质的量多于另一种单体时,聚合反应就无法再继续下去。
2)按聚合物链结构分类
线形逐步聚合反应:参加反应的单体都只带有两个官能团,聚合过程中,分子链在两个方向上增长,分子量逐步增大,体系的粘度逐渐上升,最后形成高分子的聚合反应。
支化、交联聚合反应(体型聚合):参加聚合反应的单体至少有一个含有两个以上官能团时,反应过程中,分子链从多个方向增长。调节两种单体的配比,可以生成支化聚合物或交联聚合物(体型聚合物)
3)按参加反应的单体种类分类
(1)逐步均聚反应:只有一种或两种单体参加聚合反应,生成的聚合物只含有一种重复单元。
(2)逐步共聚反应:两种或两种以上单体参加聚合反应,生成的聚合物含有两种或两种以上的重复单元。
3. 缩聚反应
缩聚反应:是缩合聚合的简称,是多次缩合重复结果形成缩聚物的过程。缩合和缩聚都是基团间的反应,两种不同基团可以分属于两种单体分子,也可能同在一种单体分子上。
官能度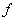 (Functionality):一分子聚合反应原料中能参与反应的官能团数称为官能度。
(Functionality):一分子聚合反应原料中能参与反应的官能团数称为官能度。
1-1、1-2、1-3体系缩合,将形成低分子物;2-2或2-官能度体系缩聚,形成线形缩聚物;2-3、2-4或3-3体系则形成体形缩聚物。
4. 线形缩聚机理
线形缩聚机理的特征有:逐步和可逆。
1)线型缩聚反应的逐步性
缩聚大分子的生长是由于官能团相互反应的结果。缩聚早期,单体很快消失,转变成二聚体、三聚体、四聚体等低聚物,转化率很高,以后的缩聚反应则在低聚物之间进行。缩聚反应就是这样逐步进行下去的,聚合度随时间或反应程度而增加。延长聚合时间的主要目的在于提高产物相对分子质量,而不在于提高转化率。缩聚早期,单体的转化率就很高,而相对分子质量却很低。
转化率:是指转变成聚合物的单体部分占起始单体量的百分数。
逐步特性是所有缩聚反应所共有的。
2)线型缩聚反应的平衡性
许多缩聚反应是可逆的,其可逆的程度可由平衡常数来衡量。根据其大小,可将线型缩聚大致分成三类:①平衡常数小,如聚酯化反应,K≈4,低分子副产物水的存在对聚合物相对分子质量影响很大,应除去。②平衡常数中等,如聚酰胺化反应,K≈300~500,水对聚合物相对分子质量有所影响。③平衡常数很大或看作不可逆,如聚碳酸酯和聚砜一类的缩聚,平衡常数总在几千以上。
可逆平衡的程度则各类缩聚反应有明显的差别。
3) 线型缩聚反应的平衡常数
Flory等活性理论:单官能团化合物的分子链达到一定长度之后,其官能团的化学反应活性与分子链长无关。
按照官能团等活性理论,可以用一个平衡常数表征整个聚合反应的平衡特征,并以体系中的官能团浓度代替单体浓度。以聚酯反应为例,则其平衡常数为
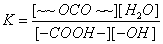
方括号的含义是代表官能团的浓度和小分子的浓度。
Flory等活性理论的适用条件:①缩聚反应体系必须是真溶液,均相体系,全部反应物、中间产物和最终产物都溶于这个介质。②官能团所处的环境——邻近基团效应和空间阻碍两方面因素在反应过程中应当不变。③聚合物的相对分子质量不能太高,反应速率不能太大,反应体系黏度不能太高,以不影响小分子产物的逸出、不妨碍建立平衡为限,不能使扩散成为控制速率的主要因素。
4) 反应程度和聚合度
考虑到在线型缩聚反应中实际参加反应的是官能团而不是整个单体分子,所以通常采用已经参加了反应的官能团与起始官能团的物质的量之比即反应程度 来表征该反应进行的程度:
来表征该反应进行的程度:
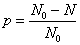 =已反应官能团数/起始官能团总数
=已反应官能团数/起始官能团总数
式中: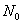 为反应起始时单体的总物质的量;N为缩聚反应体系中同系物(含单体)的总物质的量。
为反应起始时单体的总物质的量;N为缩聚反应体系中同系物(含单体)的总物质的量。
线型平衡缩聚物的数均聚合度与反应程度的关系为
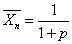
线型平衡缩聚物的重均聚合度与反应程度的关系为
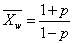
线型平衡缩聚物相对分子质量分散度为
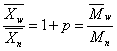
当线型平衡缩聚反应程度很高(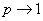 )时,聚合物的分散度接近于2。
)时,聚合物的分散度接近于2。
5) 缩聚反应中的副反应
缩聚通常在较高的温度下进行,往往伴有基团消去、化学降解、链交换等副反应。缩聚反应中的副反应:①链裂解反应是发生于缩聚物分子链与小分子有机或无机化合物之间的副反应,如聚酯的水解、醇解、酸解、胺解等。②链交换反应发生于两个大分子链之间的副反应。③环化反应是发生于大分子链内的副反应。④官能团分解反应是发生于大分子链内的副反应,如高温下羧基的脱羧、醇羟基的氧化反应等。
缩聚副反应的结果:①链裂解使聚合度降低。②链交换使分散度降低,链交换反应在一定程度上对改善缩聚物的性能有利。③环化反应使聚合反应无法进行。④官能团分解反应危及聚合反应的顺利进行。
减少缩聚副反应所采取的措施:①为了减轻链裂解副反应的影响,必须首先考虑提高原料单体的纯度,来尽可能降低有害杂质特别是单官能团化合物的含量。②提高单体浓度等有利于双(多)分子之间反应的条件可以抑制环化副反应的发生;适当降低反应温度对于减轻环化副反应的影响有一定效果。③由于官能团分解反应的活化能高于聚合反应,所以应尽可能避免反应温度过高和反应器的局部过热,同时惰性气体排除反应器中的空气是减少官能团分解副反应的有效措施。
6) 线型平衡缩聚反应的影响因素
温度、压力、单体浓度、催化剂、搅拌和惰性气体保护是影响缩聚反应的六个外因;平衡常数是影响缩聚反应的内因。
(1)反应温度的影响。①升高温度使平衡常数和聚合度降低。②升高温度会提高线型平衡缩聚反应的速率,降低体系黏度,有利于排除小分子。③升高温度会导致副反应的发生,所以必须通过试验确定最佳的反应温度。
(2)反应器内压力。①在聚合反应后期减压有利于排除小分子。②在反应初期减压不利于维持低沸点单体的等物质的量配比。所以,采取反应初期加压反应后期减压的方法,就能兼顾既不破坏原料单体的物质的量配比,又可以达到更高的反应程度和聚合度的目的。
(3)催化剂。催化剂可提高聚合反应速率,而反应平衡常数不改变。
(4)单体浓度。高的单体浓度可以得到较高相对分子质量的聚合物。
(5)搅拌。①有利于反应物料的均匀混合与扩散。②强化传热过程以利于温度控制。③有利于排除生成的小分子副产物。④高强度的搅拌剪切力可导致线形大分子链断裂,从而引发机械降解。
(6)惰性气体。①避免氧化反应的发生。②有利于排除反应过程中生成的小分子。③又可能带出单体,不利于维持低沸点单体的等物质的量配比。所以如果原料单体的沸点较低,则不宜在反应初期,而只能在反应中后期通入惰性气体。
7) 获得高相对分子质量缩聚物的基本条件
获得高相对分子质量缩聚物的重要条件是:①单体纯净,无单官能团化合物。②官能团等物质的量配比。③尽可能高的反应程度,包括温度控制、催化剂、后期减压排除小分子、惰性气体保护等。
2.2官能团等活性概念
官能团等活性概念:反应物的两个官能团的反应活性是相等的,它与分子链的大小(分子量)无关,与另一个官能团是否已经反应也无关。
适合缩聚反应的单体必须具备两个基本条件:①带有两个不同或相同的官能团。②这两种官能团之间或者与别的单体的官能团之间可以进行化学反应并生成稳定的共价键。
单体活性的三个决定因素:①官能团取代负电性。如羧酸衍生物的活性取决于酰基取代基的电负性大小,其酰基取代基的电负性越大,羧酸衍生物的活性越高。②官能团邻近基团。如甘油参加一般缩聚反应时伯羟基的反应活性较高,而促羟基的活性较低。③碳原子数及环化倾向。特别注意的是,四五个碳原子的氨基酸和羟基酸具有强烈的环化倾向而不能聚合。
2.3逐步聚合反应动力学
在二元酸和二元醇的缩聚反应中,根据Flory等活性理论,可假定每一步的速率常数相等。
1.聚酯反应机理
Flory认为酸催化是酯化反应的必要条件。原料羧酸本身是能够离解并提供质子的催化剂,发生“自催化作用”,也可以采用外加酸作催化剂。
2.聚酯反应动力学方程
参加反应的官能团是等物质的量配比时,外加酸催化的聚酯反应属于二级反应,其动力学方程为
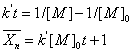
式中: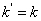 [外加酸];
[外加酸];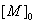 为羟基或羧基浓度。
为羟基或羧基浓度。
参加反应的官能团是等物质的量配比时,自催化三级反应动力学方程为
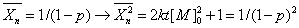
注意:上述两个动力学方程并没有考虑到逆反应。
2.4聚合度与平衡常数的关系
官能团等活性和等物质的量配比时,线型平衡缩聚反应达到平衡时聚合物同系物(其中含单体)的平均聚合度()与平衡常数(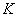 )、反应程度()以及体系中小分子存留率(
)、反应程度()以及体系中小分子存留率(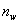 )之间的关系为
)之间的关系为

这是一个普遍公式,式中: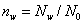 ,定义为存留在体系中小分子的物质的量分数;为生成小分子(这里用H20代表)的物质的量。
,定义为存留在体系中小分子的物质的量分数;为生成小分子(这里用H20代表)的物质的量。
1)密闭体系
平衡聚合反应:单体与聚合物之间存在平衡关系的聚合反应称为~或可逆聚合反应。通常将逆反应叫做解聚反应。
缩聚反应在与外界完全无传质过程的所谓“密闭反应器”中进行。
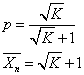
所以,密闭体系中进行的线型平衡缩聚反应达到平衡时的聚合物同系物的聚合度完全由平衡常数决定。
2)敞开体系
缩聚反应在能够与外界进行传质过程的敞开反应器中进行,即将小分子副产物不断从反应体系中移走。
当聚合物平均相对分子质量在10 000以上时,反应程度可近似地取为1,则
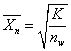 (许尔兹公式)
(许尔兹公式)
所以,对于绝大多数线型平衡缩聚反应而言,要获得高相对分子质量的聚合物就必须保证反应在敞开的反应器中进行,同时需要排出小分子副产物,使残留在反应体系中的小分子尽可能小。
2.5 线型聚合反应的分子量控制
根据不同的用途、在不同的场合对聚合物的相对分子质量控制的目的为以下二者之一:①使聚合物的相对分子质量达到或接近预期的数值。使聚合反应在达到要求的相对分子质量时失去进一步聚合的条件。可采用控制两种官能团的配比或加入端基封锁剂的方法。②使聚合物的相对分子质量尽可能高。创造使大分子两端的官能团能够无限制地进行聚合反应的条件。控制分子量通常有以下方法:①控制反应程度。②控制反应官能团的当量比。③加入少量单官能团单体。
1.控制反应程度
在任何情况下,缩聚物的聚合度均随反应程度的增加而增加。逆反应和原料非等物质的量比均使反应程度有所限制,难以获得高相对分享质量的缩聚物。
2.缩聚平衡对聚合度的影响
对于聚酯化一类可逆缩聚反应,平衡常数对反应程度进而对聚合度将产生很大影响。密闭体系中聚合度与平衡常数的定量关系为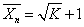 。敞开体系中聚合度与平衡常数和存留在体系中小分子的摩尔分数的定量关系为
。敞开体系中聚合度与平衡常数和存留在体系中小分子的摩尔分数的定量关系为 ,如不及时除去小分子副产物,由于逆反应,将得不到很高的反应程度和聚合度。
,如不及时除去小分子副产物,由于逆反应,将得不到很高的反应程度和聚合度。
3.线型缩聚物聚合度的控制
反应程度和平衡条件是影响线型缩聚物聚合度的重要因素,却不能用作控制的方法。控制的方法往往是在两官能团等物质的量的基础上,使某官能团(或单体)稍过量或另加少量单官能团物质,使端基封锁,不再反应,反应程度被稳定在某一数值上,就可以制得预定聚合度的产物。
1)2-2体系基团数(化学计量)不相等
双官能团单体A-A和B-B物质的量分别为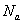 和
和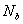 ,分别为两种单体分子数的2倍。定义
,分别为两种单体分子数的2倍。定义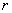 为两官能团物质的当量系数(摩尔系数)(是数值小的官能团物质的量与数值大的官能团物质的量之比)。工业上用过量分率
为两官能团物质的当量系数(摩尔系数)(是数值小的官能团物质的量与数值大的官能团物质的量之比)。工业上用过量分率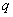 (线形缩聚中某一单体过量的摩尔分率)表示。
(线形缩聚中某一单体过量的摩尔分率)表示。
即:
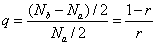
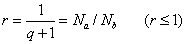
总的单体数为: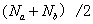 或
或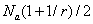
 (起始物质的总数)/(聚合物分子总数)
(起始物质的总数)/(聚合物分子总数) 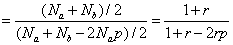
当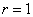 (两官能团等当量)时
(两官能团等当量)时 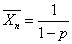
当p=1(聚合反应100%完成)时 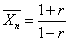
2)两双官能团单体等当量比

3)官能团A和B总是以等当量存在,即r=1,加入单官能团单体,以达到控制和稳定聚合物分子量的目的。

2.6 线型聚合反应中的分子量分布
线型平衡缩聚反应中聚合物同系物组成的摩尔分数(或数量分数)分布函数(Flory分布)为
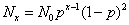
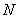 是聚合物分子总数。
是聚合物分子总数。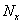 为x-聚体的数目。为起始结构单元的总数。
为x-聚体的数目。为起始结构单元的总数。
线型平衡缩聚物分子组成的质量分布函数为
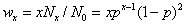
Flory分布函数的用途:①表征聚合物的相对分子质量分布;②计算任何反应程度时任何聚合度同系物的摩尔分数。
2.7 逐步聚合反应实施方法
欲使逐步聚合成功,必须考虑下列原则和措施:
①原料要尽可能纯净。
②单体按化学计量配制,加微量单官能团物质或某双官能团单体微过量来控制分子量;
③尽可能提高反应程度;
④采用减压或其他手段去除副产物,使反应向聚合物方向移动。
融熔缩聚(Melt Poly-condensation):熔融缩聚是指反应温度高于单体和缩聚物的熔点,反应体系处于熔融状态下进行的反应。熔融缩聚的关键是小分子的排除及分子量的提高。优点:①体系中组分少,设备利用率高,生产能力大。②反应设备比较简单,产品比较纯净,不需要后处理。缺点:①要求生产分子量高的聚合物时有困难。②长时间高温加热会引起氧化降解等副反应;③要求官能团物质的量比例严格,条件比较苛刻。④当聚合物熔点不超过300时,才能考虑采用熔融聚合。
溶液缩聚(Solution Poly-condensation):单体加适当催化剂在溶剂(包括水)中呈溶液状态下进行的缩聚叫溶液缩聚。特点:①反应温度较低,一般为40~100。②反应设备简单。③由于溶剂的引入,设备利用率低,由于溶剂的回收处理,使工艺过程复杂化。
选用溶剂时需要考虑的因素:①溶剂的极性。②溶剂化作用。③溶剂的副反应。
界面缩聚(Interfacial Poly-condensation):两单体分别溶解于两不互溶的溶剂中,反应在两相界面上进行的缩聚称之为界面缩聚,具有明显的表面反应的特性。
特点:①复相反应,将两单体分别溶于互不相溶的溶剂中。②不可逆。③界面缩聚的总速率决定于扩散速率。高分子量聚合物的生成与总转化率无关。④相对分子质量对配料比敏感性小。⑤反应温度低,相对分子质量高。⑥所用设备体积大,利用率低。
固相缩聚:是在玻璃化温度以上,熔点以下的固态所进行的缩聚反应。
2.8 非线型逐步聚合反应
1. 支化型逐步聚合反应
当体系存在大于两个官能团的单体时(官能度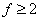 时),得到支化高分子,而不会产生交联。其中,
时),得到支化高分子,而不会产生交联。其中, 体系生成超支化高分子(hyperbranched polymer)。当超支化高分子中所有的支化点的官能度相同,且所有支化点间的链段长度相等时,叫树枝形高分子(dendrimer)。
体系生成超支化高分子(hyperbranched polymer)。当超支化高分子中所有的支化点的官能度相同,且所有支化点间的链段长度相等时,叫树枝形高分子(dendrimer)。
2. 交联型逐步聚合反应
在A-B单体与 单体(
单体( )的聚合反应体系中,若加入B-B型单体时,两个聚合物分子链之间就可以发生反应,生成交联型聚合物。这种大分子之间成键生成交联聚合物的反应称做交联反应。聚合体系中单体的平均官能度、官能团物质的量的比及反应程度,决定了聚合反应是生成支化高分子还是交联高分子(体型聚合物)。
)的聚合反应体系中,若加入B-B型单体时,两个聚合物分子链之间就可以发生反应,生成交联型聚合物。这种大分子之间成键生成交联聚合物的反应称做交联反应。聚合体系中单体的平均官能度、官能团物质的量的比及反应程度,决定了聚合反应是生成支化高分子还是交联高分子(体型聚合物)。
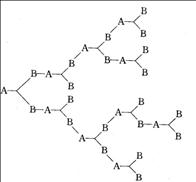
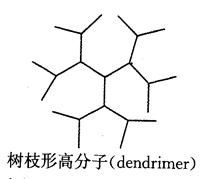
体型聚合物(热固性聚合物)在性能上具有不溶、不熔和机械强度高的特点。而线形聚合物或支链形聚合物(热塑性聚合物)则可熔融塑化,受热后,潜在官能团进一步交联而固化。
1)体型缩聚反应的特点
体型缩聚反应的特点:①可分步进行。②存在凝胶化过程。③凝胶点之后,聚合反应速率较同类线型反应的反应速率低。
凝胶化过程也叫凝胶化现象(gelation),即体型缩聚反应当反应程度达到某一数值时,反应体系的黏度会突然增加,突然转变成不溶、不熔、具有交联网状结构的弹性凝胶的过程。此时的反应程度被称做凝胶点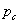 (critical reaction conversion point)。通常以气泡在体系中不能上升为判据。凝胶化过程发生时,体系中存在凝胶和溶胶两个部分。凝胶(gel)是呈交联网状结构的体形聚合物,不溶于一切溶剂;溶胶(sol)则是被包裹在凝胶的网状结构中的链形聚合物,其相对分子质量较小,是可以溶解的。溶胶可用溶剂浸取出来,溶胶还可以进一步交联成凝胶。
(critical reaction conversion point)。通常以气泡在体系中不能上升为判据。凝胶化过程发生时,体系中存在凝胶和溶胶两个部分。凝胶(gel)是呈交联网状结构的体形聚合物,不溶于一切溶剂;溶胶(sol)则是被包裹在凝胶的网状结构中的链形聚合物,其相对分子质量较小,是可以溶解的。溶胶可用溶剂浸取出来,溶胶还可以进一步交联成凝胶。
2)无规预聚物和结构预聚物
(1)无规预聚物。通常将在接近凝胶点时终止聚合反应,得到的相对分子质量不高、可以在加工成型过程中交联固化的聚合物叫做预聚物(prepolymerization)。将分子链端的未反应官能团完全无规的预聚物通常叫做无规预聚物。例如,碱催化酚醛树脂、脲醛树脂、醇酸树脂、三聚氰胺树脂(即密醛树脂)都属于此类。
在工艺上,根据反应程度的不同,将体型缩聚物的合成分为甲、乙、丙三个阶段。 甲阶树脂(A-stage resin)的反应程度小于凝胶化开始时的临界反应程度(凝胶点),甲阶聚合物具有良好的溶、熔性能。乙阶树脂(B-stage resin) 接近,溶解性能变差,但仍能熔融。丙阶树脂(C-stage resin)的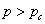 ,已经交联,不能再溶、熔。成型加工厂多使用乙阶树脂。
,已经交联,不能再溶、熔。成型加工厂多使用乙阶树脂。
(2)结构预聚物。将具有特定的活性端基或侧基、基团结构比较清楚的特殊设计的预聚物称为结构预聚物。例如,环氧树脂、不饱和聚酯树脂、酸催化酚醛树旨、制备聚氨酯用的聚醚二元醇和聚酯二元醇、遥爪聚合物都属于此类。结构预聚物往往是线型低聚物,其本身一般不能进一步聚合或交联,第二阶段交联固化时,需另加入催化剂或其他反应性物质来进行,这些加入的催化剂或其他反应物通常叫固化剂。
3. Carothers方程法
平均官能度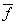 (Aver-Functionality) :是指在两种或两种以上单体参加的混缩聚或共缩聚反应中,在达到凝胶点以前的线型缩聚反应阶段,反应体系中实际能够参加反应的各种官能团总物质的量与单体总物质的量之比。
(Aver-Functionality) :是指在两种或两种以上单体参加的混缩聚或共缩聚反应中,在达到凝胶点以前的线型缩聚反应阶段,反应体系中实际能够参加反应的各种官能团总物质的量与单体总物质的量之比。
体型缩聚的重点是凝胶点计算。凝胶点计算的关键是平均官能度的计算。对于两种官能团参加的体型缩聚反应的平均官能度的计算要点是:①按照官能团的种类将单体分为两组,分别计算两种官能团的总物质的量;②比较两种官能团总物质的量的大小,判断体系官能团的配比是等物质的量还是不等物质的量,选择相应的公式计算平均官能度;③将平均官能度带入Carothers方程即可计算出凝胶点。应当注意的是计算凝胶点的数值一定小于或等于1,通常情况下应该保留三位有效数字。
Carothers对体型缩聚反应线型阶段作如下两点合理假定:①在线型缩聚阶段每进行一步反应都必然等量消耗两个不同的官能团,同时伴随着一个同系物分子的消失。②达到凝胶化过程发生的那一刻,聚合物的相对分子质量急速增大直至发生交联,此时将聚合度定义为无穷大。于是按照反应程度定义[ ,
,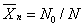 ]可以得到Carothers方程:
]可以得到Carothers方程:
(1)反应物等当量
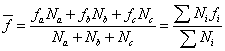
聚合度与单体平均官能度及反应程度的关系式:
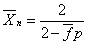
或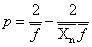
凝胶点时:
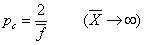
(2)反应物不等当量
两种单体非等当量时,可以简单的认为,聚合反应程度是与量少的单体有关。另一单体的过量部分对分子量增长不起作用。如对一个三元混合物体系,单体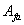 ,
,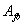 和
和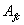 的摩尔分数分别为、和
的摩尔分数分别为、和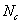 ,官能度分别为
,官能度分别为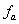 、
、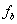 和
和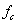 。单体和含有同样的A官能团,并且B官能团过量,即
。单体和含有同样的A官能团,并且B官能团过量,即 ,则平均官能度为:
,则平均官能度为:
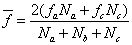
或
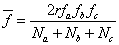
式中
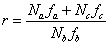
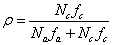
是A和B官能团的当量系数,它等于或小于1,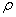 是的单体所含A官能团占总的A官能团的分数。
是的单体所含A官能团占总的A官能团的分数。
4. Flory统计凝胶点计算
支化系数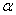 :为高分子链末端支化单元上一给定的官能团连接到另一高分了链的支化单元的几率。
:为高分子链末端支化单元上一给定的官能团连接到另一高分了链的支化单元的几率。
临界支化系数为:
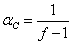
普遍情况分析:
支化系数: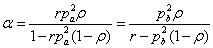
凝胶化时反应程度: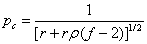
当两种官能团等当量时,r=1,且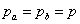 ,时:
,时:
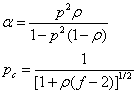
当没有A-A单体时(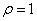 ),r<1,时
),r<1,时
在同样的反应程度下,支化聚合物的重量分布比线型聚合物的宽。当多官能团单体的官能度增大时,支化聚合物的分布变得更宽,随着的增大,分布也变宽。
5。凝胶点的测定方法
多官能团体系聚合至某一程度,体系黏度急增,气泡无法上升,此时立即取样分析残留官能团,计算反应程度,这就是实测凝胶点。通常情况下,实测凝胶点是最接近实际值。实测凝胶点的误差只是来源于实验误差。
6。三种凝胶点的数值比较
三种凝胶点的数值比较:Flory统计学公式计算的凝胶点数值< 实测凝胶点数值< Carothers方程计算的凝胶点数值。如果将Flory统计学公式和Carothers方程计算的凝胶点加以平均,则数值与实际凝胶点就比较接近。
Carothers方程计算的凝胶点数值偏小的原因:Carothers在推导凝胶点公式的时候假定聚合度无穷大时才发生凝胶化,实际上,聚合度不太高时就开始凝胶化。
Flory统计学公式计算的凝胶点数值偏大的原因:Flory在作统计学处理时未考虑分子内的环化副反应,也未考虑发生凝胶化时反应条件相对于官能团等活性条件的偏离。
反应程度(Extent of Reaction)与转化率(Conversion):参加反应的官能团数占起始官能团数的分率。参加反应的反应物(单体)与起始反应物(单体)的物质的量的比值即为转化率。
凝胶化现象(Gelation Phenomena) 凝胶点(Gel Point):体型缩聚反应进行到一定程度时,体系粘度将急剧增大,迅速转变成不溶、不熔、具有交联网状结构的弹性凝胶的过程,即出现凝胶化现象。此时的反应程度叫凝胶点。
预聚物(Pre-polymer):体形缩聚过程一般分为两个阶段,第一阶段原料单体先部分缩聚成低分子量线形或支链形预聚物,预聚物中含有尚可反应的基团,可溶可熔可塑化。该过程中形成的低分子量的聚合物即是预聚物。
无规预聚物(Random Pre-polymer):预聚物中未反应的官能团呈无规排列,经加热可进一步交联反应。这类预聚物称做无规预聚物。
结构预聚物(Structural Pre-polymer):具有特定的活性端基或侧基的预聚物称为结构预聚物。结构预聚物往往是线形低聚物,它本身不能进一步聚合或交联。
第三章自由基聚合
3.1 判断某种聚合物能否进行聚合反应
1. 可进行连锁聚合单体的特点
研究可进行连锁聚合单体的结构特点主要涉及能够作为聚合反应单体的烯烃的基本条件,以及单体结构与聚合反应类型之间的关系。具体而言,首先从烯烃取代基所造成位阻大小的角度判断其能否进行聚合,然后再从取代基电负性和共轭性的角度判断其能够进行哪一种或哪几种类型的聚合反应。
1)取代基的数目、位置、大小决定烯烃能否进行聚合
(1)一取代烯烃原则上都能够进行聚合反应。
(2)取于1,1-二取代的烯类单体,一般都能按取代基的性质进行相应机理的聚合。并且由于结构上不对称、极化程度增加,因而其更易聚合。但当两个取代基都是体积较大的芳基时,只能形成二聚体。
(3)1,2-二取代的烯类单体,由于其结构对称、极化程度低和位阻效应,一般不能均聚或只能形成二聚体。
(4)三取代乙烯和四取代乙烯一般不能聚合,但氟代乙烯是例外,不论氟代的数目和位置如何,均易聚合,这是氟的原子半径较小的缘故。
2)取代的电负性和共轭性决定烯烃的聚合反应类型
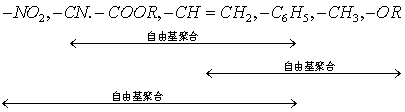
(1)带吸电子取代基的烯烃能够进行自由基型和阴离子型两种聚合反应;
(2)带推(供)电子取代基的烯烃能够进行阳离子型聚合反应。但是丙烯除外,只能进行配位聚合。
(3)带共轭取代基的烯烃能够进行自由基、阴离子和阳离子三种类型的聚合反应。
下面列出烯烃取代基的种类与其能够进行的聚合反应类型的相关性:
2. 从连锁聚合反应热力学角度分析
烯烃单体通过加成聚合反应生成聚合物的过程,是一个从无序到线形有序、熵值降低的过程。从热力学角度考虑,聚合反应过程熵值降低所造成的热力学障碍必须以分子热力学能(近似为焓增量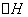 ,即聚合热的负值)的降低来补偿,也就是说聚合热越大,聚合反应越易进行。
,即聚合热的负值)的降低来补偿,也就是说聚合热越大,聚合反应越易进行。
从单体角度出发,以乙烯聚合热为基准,使聚合热改变的四个因素:①取代基位阻效应使聚合热降低;②取代基共轭效应使聚合热降低;③氢键和溶剂化效应使聚合热降低;④强电负性取代基(F,Cl)使聚合热升高。
聚合上限温度Tc (Ceiling Temperature of Polymerization):ΔG=0,聚合和解聚处于平衡状态时的温度即为聚合上限温度,在此温度以下进行的聚合反应无热力学障碍;超过聚合上限温度聚合就无法进行。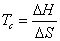 , 平衡温度:
, 平衡温度: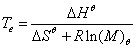
在此温度以下进行的聚合反应无热力学障碍;高于此温度聚合物将自动降解或分解;在此温度或稍低于此温度条件下单体的聚合反应十分困难。 也可以通过实验测定聚合反应转化率与温度的关系,再外推至转化率为零时的温度()。
也可以通过实验测定聚合反应转化率与温度的关系,再外推至转化率为零时的温度()。
3.2 自由基聚合反应机理
1. 自由基的产生及其活性
某些有机化合物或无机化合物中弱共价键的均裂和具有单电子转移的氧化还原反应是产生自由的两种主要方式。除此之外,加热、光照和高能辐射等方式也可以产生自由基。
自由基的活性主要决定于三个因素,即共轭效应、诱导效应和空间位阻效应。诱导效应(Induction Effect):单体的取代基的供电子、吸电子性。共轭体系(Resonance System):在某些有机化合物分子中,由于双键、p电子或空的p轨道的相互影响与作用,使得电子云不能仅仅局限在某个碳原子上,而是分散在一定范围内多个原子上的离域体系中,这种离域体系就是共扼体系。共轭效应(Resonance Effect):共扼效应存在于共扼体系中,它是由于轨道相互交盖而引起共扼体系中各键上的电子云密度发生平均化的一种电子效应。共扼效应使体系的键长趋于平均化,体系能量降低,分子趋于稳定。可分为σ-π共轭、p-π共轭、π-π共轭、σ-p共轭。空间位阻效应(Steric Effect):由取代基的体积、数量、位置所引起的效应,它对单体聚合能力有显著的影响,但它不涉及对活性种的选择。
一般而主:①取代基的共轭效应的结果,使自由基电子云密度降低,从而降低了自由基的能量,自由基稳定性增强。②取代基诱导效应:推电子取代基的+I效应使自由基电子云密度增加,能力升高,自由基稳定性降低;而吸电子取代基的-I效应使自由基电子云密度降低,能量降低,自由基稳定性增强。③空间位阻效应:取代基的位阻和排斥作用,给自由基的反应增加了困难,自由基稳定性增强。
当①和②对自由基稳定性影响发生矛盾时,共轭效应起主导作用。当②和③对自由基稳定性影响发生矛盾时,空间位阻效应起主导作用。
判断哪一类活性的自由基适合引发烯烃单体进行聚合的总原则是:①活性太高的自由基(如氢自由基或甲基自由基)的产生需要很高的活化能,自由基的产生和聚合反应的实施都相当困难。②活性太低的自由基(如卞基自由基和烯丙自由基)的产生非常容易,但是它们不仅无法引发单体聚合,反而常常会与别的活泼的自由基进行独电子之间的配对成键,形成稳定的化合物。③中等活性的自由基(如 等)和苯基自由基是引发单体进行聚合反应最常见的自由基。
等)和苯基自由基是引发单体进行聚合反应最常见的自由基。
2. 自由基聚合的基元反应
自由基聚合反应包括:链引发、链增长和链终止。链引发(Chain Initiation):形成单体自由基活性种的反应。链引发包括两步:初级自由基的形成(即引发剂的分解,吸热反应),单体自由基的形成(放热反应)。链增长(Chain Propagation):单体自由基形成后,它仍具有活性,能打开第二个烯类分子的π双键,形成新的自由基,新自由基的活性并不随着链段的增加而衰减,与其它单体分子结合成单元更多的链自由基,即链增长。其有两个特征:一是放热反应,二是增长活化能低,增长速率极高。链终止(Chain Termination):自由基活性高,有相互作用终止而失去活性的倾向。 链自由基失去活性形成稳定聚合物的反应称为链终止反应 。
自由基聚合反应的特点是:慢引发、快增长、速终止,三者的速率常数递增。其中链引发反应速率主要是由引发剂分解速率决定。链终止反应包括双基终止和转移终止两种类型。单基终止(Mono-radical Termination):链自由基从单体、溶剂、引发剂等低分子或大分子上夺取一个原子而终止,这些失去原子的分子可能形成新的自由基继续反应,也可能形成稳定的自由基而停止聚合。双基终止(Bi-radical Termination):链自由基的独电子与其它链自由基中的独电子或原子作用形成共价键的终止反应。
双基终止包括双基偶合终止和双基歧化终止。偶合终止(Coupling Termination):两链自由基的独电子相互结合成共价键的终止反应,偶合终止的结果是大分子的聚合度为链自由基重复单元数的两倍。 歧化终止(Disproportionation Termination):某链自由基夺取另一自由基的氢原子或其他原子终止反应。歧化终止的结果是聚合度与链自由基的单元数相同。
双基偶合终止有三个特点:①相对分子质量2倍于链自由基。②带2个引发剂残基。③分子中含一个头-头连接结构单元。双基歧化终止也有三个特点:①相对分子质量与链自由基相等。②带1个引发剂残基。③一半分子链端饱和,一半分子链端含双键。这各自的三个特点可用来判断某一聚合反应各终止方式所占的比例。不同单体的聚合反应具有不同的链终止反应方式,按哪种链终止方式主要取决于单体结构和反应条件。最常见的聚苯乙烯和聚丙烯腈是按双基偶合方式终止;聚甲基丙烯酸甲酯主要是按双基歧化方式终止。由于双基歧化终止涉及活化能较高的氢原子转移,所以升高温度往往会导致歧化终止倾向的增加。
链转移(Chain Transfer):在自由基聚合过程中,链自由基可能从单体(M)、溶剂(S)、引发剂(I)等低分子或大分子上夺取原子而终止,使失去原子的分子成为自由基,继续新链的增长,这一反应叫链转移反应。链转移结果,自由基数目不变。链转移反应包括:向单体转移、向引发剂转移、向溶剂转移、向大分子转移和向阻聚物质转移。
3. 自由基聚合反应特点
自由基聚合的反应特点为:①微观上,自由基聚合反应可以明显地区分成链的引发、增长、终止、转移等基元反应。其中引发速率小,是控制总聚合速率的关键。可以概括为慢引发、快增长、速终止。②只有链增长反应才使聚合度增加。一个单体分子转变成大分子的时间极短,反应不能停留在中间聚合度阶段,反应混合物仅由单体和聚合物组成。在聚合过程中,聚合度变化较小。③在聚合过程中,单体浓度逐渐降低,聚合物浓度相应提高。延长聚合时间主要是提高转化率,对相对分子质量影响较小。④少量(0.01%~0.1%)阻聚剂足以使自由基聚合反应终止。
3.3 链引发反应
烯类单体可采用引发剂产生活性种、引发聚合。在某些特殊情况下,也可采用热、光、高能辐射等引发方式。引发剂(Initiator):在聚合体系中能够形成活性中心的物质,使单体在其上连接分为自由基引发剂,离子引发剂。
1. 引发剂和引发作用
1)引发剂种类
常用的自由基聚合反应引发剂包括过氧类化合物、偶氮类化合物以及氧化还原反应体系三大类。过氧人苯甲酰(BPO)、偶氮二异丁腈(AIBN)、过硫酸盐、亚铁离子与过氧化氢(含其他过硫酸盐)的氧化还原体系是最重要的四种引发剂。其中BPO和AIBN是油溶性引发剂,过硫酸盐是水溶性引发剂。值得一提的是,AIBN分解后形成的异丁腈自由基是碳自由基,缺乏脱氢能力,因此不能用作接枝聚合的引发剂。氧化还原引发体系的优点是活化能较低,可在较低温度(5~50℃)下引发聚合,而且具有较高的聚合速率。氧化还原引发体系的组分可以是无机化合物或有机化合物,其性质可以是水溶性或油溶性。
过氧化物和偶氮化合物可以经热分解产生自由基,也可以在光照条件下分解产生自由基。
2)引发剂分解反应动力学
通常用半衰期(Half Life):物质分解至起始浓度(计时起点浓度)一半时所需的时间。
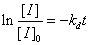
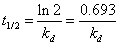
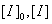 分别表示引发剂起始(t=0)浓度和t时的浓度,单位为mol·L-1,实验中只需测定恒定温度条件下引发剂浓度与时间的对应变化关系,以
分别表示引发剂起始(t=0)浓度和t时的浓度,单位为mol·L-1,实验中只需测定恒定温度条件下引发剂浓度与时间的对应变化关系,以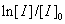 对t作图,便可求得
对t作图,便可求得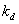 。
。
引发剂速率常数与温度关系遵循Arrehenius经验公式:
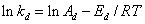
在不同温度下,测得某一引发剂的多个分解速率常数,以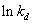 对1/T作图,由截距可求得频率因子
对1/T作图,由截距可求得频率因子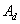 ,由斜率可求出分解活化能
,由斜率可求出分解活化能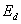 .
.
3) 引发效率
引发效率:是指引发剂分解生成的初级自由基总量中真正能够与单体反应最后生成单体自由基并开始链增长反应的百分数。造成降低的主要因素是引发剂的诱导分解和溶剂的笼蔽效应。
引发剂效率(Initiator Efficiency):引发聚合部分引发剂占引发剂分解消耗总量的分率称为引发剂效率。
诱导分解(Induced Decomposition):诱导分解实际上是自由基向引发剂的转移反应,其结果是消耗一分子引发剂而自由基数目并不增加,从而使引发剂效率降低。
AIBN一般无诱导分解。氢过氧化物ROOH特别容易发生诱导分解。丙烯腈、苯乙烯等活性较高的单体,能迅速与引发剂作用,引发增长,因此较高。相反,如乙酸乙烯酯一类低活性单体,对自由基的捕捉能力较弱,为诱导分解创造条件,因此较低。
笼蔽效应(Cage Effect):在溶液聚合反应中,浓度较低的引发剂分子及其分解出的初级自由基始终处于含大量溶剂分子的高黏度聚合物溶液的包围之中,一部分初级自由基无法与单本分子接触而更容易发生向引发剂或溶剂的转移反应,从而使引发剂效率降低。
AIBN在溶液聚合中可能发生初级自由基的双基终止而使降低。
4)引发剂选择的一般原则
引发剂的选择有四个方面:溶解类型,半衰期,特性要求,用量。
(1)按照聚合反应实施方法选择引发剂的溶解类型:对于本体聚合、悬浮聚合和一般的溶液聚合,选择油溶性引发剂如BPO、AIBN等,也可以选择油溶性的氧化还原引发体系。对于乳液聚合九以水作为溶剂的溶液聚合,宜选择水溶性引发剂如KPS,APS或水溶性氧化还原体系。
(2)按照聚合反应温度选择半衰期适当的引发剂:一般而主,引发剂在聚合反应温度下的半衰期应该与聚合反应时间处于同一数量级。例如反应温度为30~100℃时,可选择BPO、AIBN过硫酸盐等引发剂。
(3)按照聚合物的特殊用途选择符合质量要求的引发剂。如过氧类引发剂合成的聚合物容易变色而不能用于有机玻璃等光学高分子材料的合成,偶氮类引发剂有毒而不能用于与医药、食品有关的聚合物的合成。
(4)引发剂的用量一般通过实验确定:引发剂的用量大约为单体质量(或物质的量)的0.1~2%
2. 其它引发方式
热引发聚合(Thermal-Initiation Polymerization):聚合单体中不加入引发剂,单体只在热的作用下,进行的聚合称为热引发聚合。一般而言,活泼单体如苯乙烯及其衍生物、甲基丙烯酸甲酯等容易发生热引发聚合。
光引发聚合(Photo-Initiation Polymerization):单体在光的激发下(不加入引发剂),发生的聚合称为光引发聚合。可分为直接光引发聚合和光敏聚合两种。
光直接引发聚合:单体吸收一定波长的光量子后成为激发态,再分解成自由基而进行聚合反应。能直接接受光照进行聚合的单体一般是一些含有光敏基团的单体,如丙烯酰胺、丙烯腈、丙烯酸(酯),苯乙烯等。
光敏聚合:在光敏引发剂存在下,单体吸收光能而受激发,接着分解成自由基,再引发单体聚合。
光敏聚合有光敏引发剂直接引发聚合和间接光敏引发剂间接引发聚合两种。光敏引发剂直接引发聚合:光敏引发剂经光激发后,可成为自由基,进而引发单体进行的聚合反应。常用的光敏引发剂有AIBN、甲基乙烯基酮和安息香等。间接光敏引发剂间接引发聚合:间接光敏剂吸收光后,本身并不直接形成自由基,而是将吸收的光能传递给单体或引发剂而引发聚合。常用的间接光敏剂有二苯甲酮和荧光素、曙红等。
光敏剂(photosensitizer):指那些受到光照容易发生分子内电子激发的一类化合物。
光引发效率(Photo-Initiation Efficiency):又称为自由基的量子产率,表示每吸收一个光量子产生的自由基对数。
辐射聚合(Radiation Polymerization):以高能辐射线引发单体聚合,即为辐射聚合。
辐射剂量(Radiation Dosage):指辐射线传给物质的能量,一般将每克物质吸收10-5J的能量作为辐射吸收剂量的单体。辐射吸收剂量与剂量率可用于衡量辐射聚合效应的大小。
剂量率(Dose Rate):是指单体时间内的吸收剂量。
3.4 自由基聚合反应速率
1. 聚合过程
聚合过程的速率变化常用转化率-时间曲线表示。整个聚合过程一般可以分为诱导期、聚合初期、中期、后期四个阶段。
诱导期:聚合初期初级自由基不是引发单体聚合而是用于消耗体系内存在的杂质所需的时间。在诱导期内无聚合物形成,聚合速率为零。
2. 聚合反应初期动力学
四个基本假设:①忽略链转移反应,终止方式为双基终止。②Flory等活性理论:链自由基的活性与链长短无关,即各步链增长常数相等,可用kp表示。③稳定假定:在反应开始短时间后,增长链自由基的生成速率等于其消耗速率( ),即链自由基浓度保持不变,呈稳态,
),即链自由基浓度保持不变,呈稳态, 。④聚合产物的聚合度很大,链引发所消耗的单体远少于链增长过程产生的单体,因此可以认为单体仅消耗于链增长反应。
。④聚合产物的聚合度很大,链引发所消耗的单体远少于链增长过程产生的单体,因此可以认为单体仅消耗于链增长反应。


3.温度对聚合速率的影响
总速率常数k(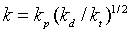 )与温度T(K)的关系遵循Arrhsnius方程式:
)与温度T(K)的关系遵循Arrhsnius方程式:
 两端取对数,则
两端取对数,则
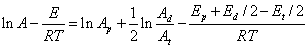
由于E为大于0的数值,所以升高温度将导致聚合速率的升高。从另一个角度讲,选择高活性(低活化能)引发剂,同样能够提高聚合速率。如采用低活化能的氧化还原引发体系能够在较低温度下获得较高的聚合速率。
4. 自动加速现象
自动加速现象(Auto-accelerative Phenomena):聚合中期(聚合反应的转化率达到15~20%以上时)随着聚合的进行,聚合速率逐渐增加,出现自动加速现象,自动加速现象主要是体系粘度增加所引起的,因此又称凝胶效应。其产生和发展的过程如下:粘度升高导致大分子链端自由基被非活性的分子链包围甚至包裹,自由基之间的双基终止变得困难,体系中自由基的消耗速率减小而产生速率却变化不大,最终导致自由基浓度迅速升高。其结果是聚合反应速率迅速增大,体系温度升高。这一结果又反馈回来使引发剂分解速率加快,这就导致了自由浓度进一步升高。于是形成循环正反馈:
自由加速过程产生的结果:①导致聚合反应速率的迅速增加,体系温度迅速升高。②导致相对分子质量和分散度都升高。③自动加速过程如果控制不当有可能严重影响产品质量,甚至发生局部过热,并最终导致爆聚和喷料等事故。
影响自动加速现象程度和出现早晚的因素:①聚合物在单体或溶剂中溶解性能的好坏,会影响到链自由基卷曲、包埋的程度,以致对双基终止速率的影响很大。自动加速现象在不溶解聚合物的非溶剂中出现的较早、较明显,此时可能有单基终止,对引发剂浓度的反应级数将为0.5~1,极限的情况(如丙烯腈)会接近于1。自动加速现象在良溶剂中较少出现,在不良溶剂中的情况则介于非溶剂(沉淀剂)和良溶剂之间。②温度的影响体现在温度对聚合体系粘度的影响。由于在较低温度下聚合体系的粘度较高,所以自动加速现象出现得较早、较明显。
5. 聚合速率的测定方法
聚合速率可以用单位时间内单体消耗或聚合物生成量来表示(通常用反应的转化率来检测):
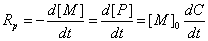 (
(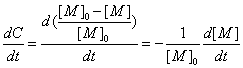 )
)
聚合速率可采用直接法和间接法来测定。直接法是用沉淀法测定聚合物量。间接法是测定聚合过程中比体积、粘度、折光率、介电常数、吸收光谱等物理性质的变化,间接求取聚合物量。常用的是经体积的测定---膨胀计法。
3.5 聚合度和链转移反应
1. 动力学链长
动力学链长(Kinetics Chain Length):每个活性种从引发阶段到终止阶段所消耗的单体分子数定义为动力学链长,动力学链在链转移反应中不终止。在自由基聚合中,增加引发剂或自由基浓度来提高聚合速率的措施,往往使产物分子量降低。引发剂引发时,产物平均聚合度一般随着温度升高而降低。在稳态、无链转移反应时,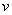 等于链增长速率与链终止速率(或引发)之比,
等于链增长速率与链终止速率(或引发)之比,
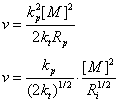 ,当引发剂引发时,引发速率
,当引发剂引发时,引发速率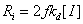 ,其方程见上表。
,其方程见上表。
2. 无链转移时的聚合度
双基偶合终止时,平均聚合度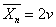 ,双基歧化终止时,
,双基歧化终止时,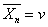 。兼有两种方式终止时,则
。兼有两种方式终止时,则 ,其值为:
,其值为:
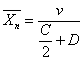 , C,D分别为偶合终止、歧化终止的分率。
, C,D分别为偶合终止、歧化终止的分率。
3. 聚合温度对聚合度的影响
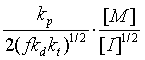
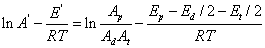
4. 链转移反应对聚合反应速率和聚合度的影响
链转移反应通常包括链自由基向单体、向引发剂、向溶剂、向大分子的转移反应。发生链转移反应的结果包括两个方面:①向单体、引发剂和溶剂的转移反应均导致链自由基的提前终止,从而使聚合度降低;向大分子的转移反应使分散度增加。②聚合反应速率的变化视链转移常数和再引发速率常数的相对大小定,如下表所示

平均聚合度:就是增长速度与形成大分子的所有终止速度(包括转移终止)之比。

(1)向单体转移
向单体转移能力与单体的结构、湿度等因素有关。键合力较小的原子(如叔氢原子、氯原子等)容易被自由基所夺取而发生链转移反应。
向单体转移的规律是:自由基的活性起决定性作用,活泼单体的自由基不活泼而不易发生转移,不活泼单体的自由基活泼而容易发生转移。
(2)向引发剂转移
自由基向引发剂转移,即链自由基对引发剂的诱导分解,使引发剂效率降低,同时也使聚合度降低。
(3)向溶剂或链转移剂转移
一般活性较大的单体(如苯乙烯)其自由基活性较小,对同一溶剂的转移常数一般要比低活性大的单体(如乙酸乙烯酯)的转移常数小。因为链增长和链移转是一对竞争反应,自由基对高活性单体反应快,链转移相对减弱。含有活泼氢或其他活泼原子如硫、氯等的溶剂容易发生转移。
(4)向大分子转移
向大分子的链转移反应往往发生于聚合反应的中后期。转移的结果是在大分子主链上形成活性中心并开始链增长,最后生成支链甚至交联。这种链自由基向别的大分子进行的所谓分子间转移反应多产生长支链;而有一种所谓分子内转移反应则多生成短支链大分子。
向大分子转移的结果:向大分子转移不改变聚合反应速率;向大分子转移产生支链的结果是使自由基型聚合物的分散度大大的提高。
链转移常数(Chain Transfer Constant):是链转移速率常数和增长速率常数之比,代表链转移反应与链增长反应的竞争能力。
链转移剂(Chain Transfer Agent):聚合物生产过程中人为地加入的一种自由基能够向其转移的试剂,用于调节聚合物分子量。常用的链转移剂有脂肪族硫醇等。
自由基捕捉剂(Radical Catcher):指1,1-二苯基-2-三硝基苯肼(DPPH)和FeCl3这两种高效阻聚剂,它们能够化学计量的1对1消灭自由基。
自由基寿命(Radical Lifetime):指自由基从产生到终止所经历的时间,可由稳态时的自由基浓度与自由基消失速率相除求得。 ,其测定方法有两种:①在光照开始或光灭以后的非稳态阶段进行。②利用光间断照射的假稳态阶段进行。
,其测定方法有两种:①在光照开始或光灭以后的非稳态阶段进行。②利用光间断照射的假稳态阶段进行。
3.6 阻聚和缓聚
阻聚剂(Inhibitor):能够使每一自由基都终止,形成非自由基物质,或形成活性低、不足以再引发的自由基的试剂,它能使聚合完全停止。按机理可分为加成型阻聚剂(如苯醌等)、链转移型阻聚剂(如DPPH等)和电荷转移型阻聚剂(如FeCl3等)等。
缓聚剂(Retarder):能够使一部分自由基终止,聚合减慢的试剂。通常不出现诱导期。
阻聚常数(Inhibition Constant):阻聚反应速率常数与增长速率常数的比值称为阻聚常数,可用来衡量阻聚效率。
烯丙基型单体的自动阻聚作用:由于链自由基容易发生向单体转移而生成稳定的烯丙基自由基,因此只能得到低聚物。
3.7 相对分子质量控制、分布及影响因素
1. 相对分子质量控制及影响因素
影响聚合度的因素包括:①单体纯度和浓度,提高单体浓度能够同时提高聚合反应速率和聚合度;提高单体浓度有利于减轻各种链转移反应对聚合度的负面影响。因此,如果希望得到尽可能高的聚合度,必须选择除单体和引发剂以外不加任何溶剂的本体聚合或悬浮聚合方法,以保证尽可能高的单体浓度。②引发剂活性和浓度,如果希望得到尽可能高的聚合度,必须控制较低的引发剂浓度。不过如果引发剂尝试太低可能使聚合反应速率太慢甚至不能进行。除此之外,选择不容易发生诱导分解的引发剂(如AIBN)对于提高聚合度是有利的。③温度,聚合度一般随着温度升高而降低。温度对大分子微观结构的影响:(a)温度升高有利于支链的生成。(b)温度升高有利于大分子链上结构单元的头-头连接。(c)温度升高有利于顺式异构体的生成。(d)降低温度有利于稳定性较好的间同立构结构单元的生成。④聚合反应方法。
2. 相对分子量分布
成键几率:是增长速率与增长和终止速度和之比(无链转移时)。

 (歧化终止)
(歧化终止)
 (偶合终止)
(偶合终止)
活性种(Reactive Species):打开单体的π键,使链引发和增长的物质,活性种可以是自由基,也可以是阳离子和阴离子。
均裂(Homolysis):化合物共价键的断裂形式,均裂的结果,共价键上一对电子分属两个基团,使每个基团带有一个独电子,这个带独电子的基团呈中性,称为自由基。
异裂(Heterolysis):化合物共价键的断裂形式,异裂的结果,共价键上一对电子全部归属于其中一个基团,这个基团形成阴离子,而另一缺电子的基团,称为阳离子。
自由基聚合(Radical Polymerization):以自由基作为活性中心的连锁聚合。
离子聚合(Ionic Polymerization):活性中心为阴、阳离子的连锁聚合。
阳离子聚合(Cationic Polymerization):以阳离子作为活性中心的连锁聚合。
阴离子聚合(Anionic Polymerization):以阳离子作为活性中心的连锁聚合。带有供电基团(如烷氧基、烷基、苯基、乙烯基等),使碳-碳双键电子云密度增加,有利于阳离的进攻和结合.而腈基和羰基(醛、酮、酸、酯)等吸电子基团使双键电子云密度降低,并使阴离子增长种共轭稳定,故有利于阴离子聚合。
转化率(Conversion):单体转化为聚合物的分率,等于转化为聚合物的单体量比去用去单体总量。
聚合动力学(Kinetics of Polymerization):指聚合速率、分子量与引发剂浓度、单体浓度、聚合温度等因素间的定量关系。