XX中药保护延长保护期的临床试验研究委托合同
甲方:
乙方:
XXX公司(以下简称“甲方”) 为合法的药品生产企业。XXXXX有限公司(以下简称“乙方”)为合法的药品临床试验验证代理公司。依据《中华人民共和国合同法》、就甲方委托乙方完成XXXX中药保护延长保护期的临床试验研究、临床试验组织,临床试验资料完善整理、临床试验总结提供相关的技术服务,双方经过平等协商,在真实、充分表达意愿的基础上,达成以下协议,并由双方共同恪守。
一、双方承担的责任和义务:
(一)甲方责任和义务:
1、在本合同签订后一周内,向乙方提供开展临床试验工作必须提供的甲方营业执照、生产许可证、GMP证书、药品批件、质量标准、药品说明书、产品物价单、药品检验报告共8个文件,并保证以上资料的真实性,且符合国家中保审评要求。
2、根据临床试验方案向乙方(按照方案规定数量)无偿提供合格的临床试验用药品
3、按合同规定的付款方式,向乙方支付研究经费。
4、根据临床试验进度,甲方定期向乙方派出监察员监察临床相关工作。
5、 甲方保存抗感胶囊中药保护延长保护期的临床研究相关资料以及临床研究所有的合法文件和原始记录。
(二)乙方责任和义务:
根据《药物临床试验质量管理规范》(GCP)、《中药品种保护条例》及国家食品药品监督管理局中药保护品种首保和续保的有关规定,在规定的临床试验期限内完成临床试验的组织、协调、监查、数据管理与统计、提交临床研究资料等临床研究工作;完成该品种临床试验相对应的适应症和病例数并符合国家食品药品监督管理局颁布的该品种续保的有关临床研究的要求。具体如下:
1、向甲方提供乙方相应的合法资质,包括营业执照、组织机构代码及税务登记证(盖乙方鲜章)。
2、负责确定临床试验参加医院和牵头单位,与医院方面签订临床研究合同,支付研究费用。本次组织参与试验的临床医院应符合新的“中药品种审评技术指导原则”的规定。临床试验负责单位应为国家药物临床试验机构,参与临床试验工作的医院资质均为三级甲等医院。
2、负责向甲方提供临床试验医院的相关资质和临床试验方案。
3、负责制订、印刷临床研究方案、病例报告表、知情同意书。
4、负责任命经GCP培训的监查员对临床研究进行定期的监查;确保临床试验遵守临床试验方案和相关法规要求;确保试验资料完整、规范并可溯源。
5、在甲方保证提供给乙方的该药品的质量标准和说明书符合国家中保审评要求的前提下,确保该临床研究资料能通过国家中药品种保护审评委员会及有关主管部门对中药保护品种临床试验审评和临床现场核查。
6. 负责协调中保办和中保审评专家的政府事务,确保甲方取得中保证书。
7、对合作过程中了解或知悉的甲方技术秘密与商业秘密承担保密义务,对抗感胶囊临床研究资料及有关数据承担保密义务,如有泄露甲方有权要求乙方承担因此造成的一切损失。
8、如果试验过程中有严重不良事件,乙方应及时配合医院处理,并及时(含观察不超过24小时)报告甲方,同时协助处理相关事项。
9、负责申报资料有关临床研究部分的补充、修改、完善,且甲方不再另行支付费用。
10、向甲方提供临床研究全套申报资料及其相应的电子文件,包括牵头单位及参加医院资质证明、临床试验方案、各医院小结表、统计分析报告、临床试验总结报告,以上资料一式六份,并加盖临床研究单位公章,且临床研究资料应有主要研究者和研究负责人的亲笔签名。
11、向甲方提供临床研究所有的合法文件原件和原始记录,主要包括编盲记录、盲态审核报告、揭盲记录、临床监察报告、知情同意书、病例报告表、发药登记表、各医院仪测仪器及正常检测值范围表、受试者入选登记表、主要研究者签名样张等,以供主管部门的临床现场核查及甲方存档。
二、临床试验期限:
乙方应在该合同签订后并在首款(合同签订的七个工作日内)和试验药物(合同签订后的十个工作日内)及其相关资质材料全部到位之日起,于XX年X月X日前完成临床试验研究,并将抗感胶囊中药保护延长保护期的临床试验资料(参加医院资质证明、临床试验方案、各医院小结表、统计分析报告、临床试验总结报告一式六份)交与甲方。
三、合同费用、支付时间和方式:
甲方付给乙方技术服务费总金额为:XXX万元(人民币XXXXXXX)。具体支付方式如下:
1、 在合同签订七个工作日内,甲方向乙方支付临床试验费总额的25%, XXXX万元(XXXXXXXX)人民币的前期工作费用;
2、 在乙方向甲方提供甲方、乙方、医院签定的三方协议和临床试验方案后的五个工作日内,甲方向乙方支付临床试验总费用的20%, XXXX万元(XXXXXX元)人民币;(累积为总费用45%);
3、 乙方将全套临床试验申报资料提供给甲方并经中保办受理后的五个工作日内,甲方向乙方支付临床试验总费用的20%,XXXXX万元(XXXX元)人民币,(累积为总费用的65%);
4、 在乙方通过宁夏区药监局的临床试验现场核查后的五个工作日内,甲方向乙方支付临床试验总费用的20%,12.52万元(壹拾贰万伍仟贰佰元)人民币,(累积为总费用的85%);
5、 甲方收到中药保护证书后七个工作日内支付剩余的乙方临床试验总费用的15%, XXXXX万元(XXXXX元)人民币。
甲方支付每一笔款后,乙方应在一个月内向甲方出据金额相等的发票。
四、合同执行与赔偿:
1、由于乙方提供的临床研究申报资料不能通过中药保护品种审评,导致甲方抗感胶囊中药保护续保申请失败,乙方应承担全部责任,并按合同总金额赔偿甲方(将甲方支付给乙方的所有临床费用退还给甲方)。
2、中药保护品种评审中如发生补充临床研究资料的情况,乙方在相关部门要求补充资料期限的时间内完成临床资料补充的工作。
3、乙方临床试验研究时间若超过临床研究完成时限,其超出时间按每天2000元向甲方支付违约金,违约金可在未支付的尾款中扣除。
4、在临床试验中,如果出现严重不良反应,乙方应及时报告申办方(含观察时间24小时内),由研究方和甲乙双方协商解决不良反应的问题,费用由甲方承担。
五、临床试验研究资料及成果的所有权归属
全部归甲方所有,乙方不得私藏,如有发现,甲方有权要求赔偿。
六、合同的终至
合同执行期间遇国家政策调整和不可抗力因素影响,双方协商终至合同执行。本合同所指不可抗力因素,除法律规定情形之外,还包括以下情形:如国家政策、法规调整、重大突发流行疾病发生等原因影响医院不能正常开展的临床研究工作;国家政策、瘟疫、毁灭性地震、重大台风水灾的发生和影响,乙方为此不承担相关过错责任或赔偿责任。
七、经双方协商订立的附加条款将作为本协议的组成部分,具有同等的法律效力。
八、争议的解决办法:
本合同条款和未及条款如发生争议,双方友好协商解决,如协商不成,可向甲方所在地人民法院提起诉讼。
九、其他:
本合同一式肆份,用中文书写,甲方执贰份,乙方执贰份,经双方单位和其法定代表人盖章签字后即生效,且每份具有同等的法律效力,自双方签字盖章之日起生效。
第二篇:药物临床试验质量管理规范
《中成藥藥品臨床試驗質量管理規範》
中成藥藥品臨床試驗質量管理規範目錄
第一章
第二章
第三章
第四章
第五章
第六章
第七章
第八章
第九章
第十章
第十一章
第十二章
第十三章
附錄一
附錄二
附錄三 總則 臨床試驗前的準備與必要條件 受試者的權益保護 試驗方案 研究者的職責 申辦者的職責 監查員的職責 紀錄與報告 數據管理與統計分析 試驗用藥的管理 質量保證 多中心試驗 研究者手冊 世界醫學大會赫爾辛基宣言 名詞釋義 臨床試驗保存文件
1
中成藥藥品臨床試驗質量管理規範
第一章
第一條
第二條
第三條
第四條
第二章
第五條
第六條
第七條
第八條
第九條
第十條
第十一條
第十二條
總則 為保證臨床試驗過程規範、結果科學可靠、保護受試者的權益並保障其安全,參照國際公認原則,制定本規範。本規範將成為中成藥臨床試驗的共同遵循的指導原則。 《中成藥藥品臨床試驗質量管理規範》是臨床試驗過程的標準規定,包括方案設計、組織、實施、監查、稽查、記錄、分析總結和報告。 凡進行各期臨床試驗,包括人體生物利用度或生物等效性試驗,均須按本規範執行。 有關一般臨床試驗的人員架構,可參閱附件1。 臨床試驗前的準備與必要條件 所有以人為對象的研究必須符合《世界醫學大會赫爾辛基宣言》的原則(附錄1),即公正、尊重病人權益、力求使受試者最大程度受益和盡可能避免傷害。參加臨床試驗的各方都必須充分了解和遵循這些原則。 進行藥品臨床試驗必須有充分的科學依據。準備在人體進行試驗前,必須周密考慮該試驗的目的及要解決的問題,應權衡對受試者和公眾健康預期的受益及可能產生的損害,預期的受益應超過可能出現的損害。選擇臨床試驗方法必須符合科學和倫理標準。 臨床試驗用藥由申辦者準備和提供。進行臨床試驗前,申辦者必須提供該試驗用藥品的臨床前研究資料。所提供的臨床前資料必須符合進行相應各期臨床試驗的要求,同時還應提供該試驗用藥品已完成和其他地區正在進行與臨床試驗有關的療效和安全性資料。試驗用藥的製造、處理及儲存應按照適用的《藥品生產質量管理規範》(GMP) 執行,並應按照已獲准的方案使用。 藥品臨床試驗機構的設施與條件必須符合安全有效地進行臨床試驗的需要。所有研究者都應具備承擔該項臨床試驗的專業特長、資格和能力。臨床試驗開始前,研究者和申辦者應就試驗方案、試驗的監查、稽查和標準操作規程以及試驗中的職責分工等達成書面協議。 臨床試驗應遵循事先已經得到倫理委員會批准的方案進行。 應由合資格的醫生或註冊中醫師負責受試者的醫學決定。 應依照相關的尊重私隱和保密規定的法例要求,來保護能夠鑒別受試者身份的紀錄。 所有臨床試驗資料的記錄、處理和儲存方式,都應允許資料可被準2
第三章
第十三條 第十四條 第十五條 第十六條 第十七條 第十八條 第十九條 確地報告、解釋和核對。 受試者的權益保護 在藥品臨床試驗的過程中,必須對受試者的個人權益給予充分的保障,應特別注意那些可能包括有弱勢對象的試驗,並確保試驗的科學性和可靠性。受試者的權益、安全和健康必須高於對科學和社會利益的考慮。倫理委員會與知情同意書是保障受試者權益的主要措施。 為確保臨床試驗中受試者的權益、安全和健康並為之提供公眾保證,應在試驗機構內成立獨立的倫理委員會,並向有關的藥品監督管理部門備案。 倫理委員會應有從事醫藥相關專業人員、非醫藥專業或非科學專業人員、法律專家及來自非試驗機構/試驗單位的人員。該會至少由五人組成,並有不同性別的委員。他們整體上都應有審評及評價有關試驗在科學,醫學及倫理方面問題的資格及經驗。倫理委員會應保存各委員的姓名及資格等紀錄。倫理委員會的組成和工作應不受任何參與試驗者的影響。 臨床試驗開始前,試驗方案需經倫理委員會審議同意並簽署批准意見後實施。在試驗進行期間,試驗方案的任何修改均經倫理委員會批准後執行,除非是研究者為消除受試者的即時危害或只涉及試驗的後勤或行政上的問題。試驗中發生任何嚴重不良事件,應及時向倫理委員會報告。 倫理委員會應獲取以下文件: 試驗方案/修改、研究者擬用於試驗的書面知情同意書及其更新件,受試者招募程序(如廣告)、提供予受試者的書面資料,研究者手冊,現有的安全性資料,受試者可獲得的款項及補償的資料,研究者的當前履歷和/或其他相關證明其資格的文件,以及其他倫理委員會履行其職務時需要的文件。 倫理委員會對臨床試驗方案的審查意見應在討論後以投票方式作出決定,參與該臨床試驗的委員不投票。倫理委員會應當在達到其書面操作程序中規定的法定人數的正式會議上作出決定。因工作需要可邀請非委員的專家或研究者出席會議及提供資料,但不投票。倫理委員會應建立其工作程序,所有會議及其決議均有書面紀錄,紀錄保存至臨床試驗結束後五年。 倫理委員會應從保障受試者權益的角度嚴格按下列各項審議試驗方案: (一) 研究者的資格、經驗、是否有充分的時間參加臨床試驗,人員配備及設備條件等是否符合試驗要求。研究者應提供當前3
的履歷或其他相關文件以證明其資格。
(二) 試驗方案是否充分考慮了倫理原則,包括研究目的、受試者
及其他人員可能遭受的風險和受益及試驗設計的科學性。
(三) 受試者入選的方法,向受試者(或其家屬、監護人、合法代
表)提供有關本試驗的信息資料是否完整易懂,獲取知情同
意書的方法是否適當。
(四) 受試者因參加臨床試驗而受到損害甚至發生死亡時,給予的
治療和/或保險措施。
(五) 試驗方案提出的修正意見是否可接受。
(六) 定期審查臨床試驗進行中受試者的風險程度,至少每年進行一次。
(七) 對支付予受試者款項的數量及方式進行審評,以確保沒有對
受試者造成強迫或不正當的影響。支付予受試者的款項應當
按比例支付,而不是完全以受試者完成試驗作為條件。倫理
委員會應當保證有關支付款項予受試者的資料,已列於知情
同意書或其他提供予受試者的書面資料之中。
第二十條 倫理委員會應在接到申請後及時召開會議,審閱討論,簽發書面意
見,並附上出席會議的委員名單、其專業情況及本人簽名。倫理委
員會的意見可以是:
(一) 批准
(二) 作必要的修正後批准
(三) 不批准
(四) 終止或暫停已批准的試驗。
第二十一條 研究者或其指定的代表必須向受試者說明有關臨床試驗的詳細情
況:
(一) 受試者參加試驗應是自願的,而且有權在試驗的任何階段隨
時退出試驗而不會遭到歧視或報復,其醫療待遇與權益不會
受到影響。
(二) 必須使受試者了解,參加試驗及在試驗中的個人資料均屬保
密。倫理委員會、有關的藥品監督管理部門或申辦者在工作
需要時,按規定可以查閱參加試驗的受試者資料。
(三) 告知受試者有關試驗的目的、試驗的過程與期限、檢查操
作、預期可能的受益和可能發生的風險與不便,他們並可能
被分配到試驗的不同組別。
(四) 必須給受試者充分的時間及機會以便考慮是否願意參加試
驗。對無能力表達同意的受試者,應向其合法代表提供上述
介紹與說明。知情同意過程應採用受試者或其合法代表能理
解的語言和文字。試驗期間,受試者可隨時了解與其有關的
4
信息資料。如果得到與受試者繼續參與試驗的願望可能相關的資料時,會及時通知受試者或其合法代表。
(五) 如發生與試驗相關的損害時,受試者可以獲得治療和相應的
補償。
(六) 受試者的責任。
(七) 預期按比例給予受試者的報酬及預計受試者因參與試驗的
支出。
(八) 對能鑒別受試者身份的紀錄應保密。當試驗結果發表時,受
試者的身份應仍然保密。
(九) 如果得到試驗的進一步資料或有關受試者權益的資料時,及
在發生與試驗相關損害時的聯絡人。
(十) 預期受試者的參與可能會被中止的情況及/或原因。 (十一) 預計的受試者人數。
第二十二條 經充分和詳細解釋試驗的情況後獲得知情同意書。
(一) 由受試者或其合法代表在知情同意書上簽署並註明日期,執
行知情同意過程的研究者也需在知情同意書上簽名並註明日期。在受試者正式參加試驗之前,受試者或其合法代表應收到一份已簽署並註明日期的書面知情同意書的副本及其他提供予受試者的書面資料。
(二) 對無行為能力的受試者,如果倫理委員會原則上同意、研究
者認為受試者參加試驗符合其本身利益時,則這些病人也可以進入試驗,同時應由其合法代表簽署並註明日期。
(三) 如果受試者或其合法代表無閱讀能力,在整個知情同意討論
期間必須有一名中立的見證人在場。在書面的知情同意書和其他文字資料交給受試者後,研究者或其指定代表須向受試者或其合法代表進行解釋,若受試者或其合法代表已經口頭同意參加試驗,在可能的情況下在知情同意書上親自簽署並註明日期,而見證人亦應親自在知情同意書上簽署並註明日期。通過見證人在知情同意書上簽署,證明有關知情同意書及其他書面資料已經準確地向受試者或其合法代表解釋,而受試者或其合法代表也已完全明白,同時該知情同意是受試者或其合法代表自願給予的。
(四) 兒童原則上不能作為受試者,除非試驗用藥品擬定的主治病
證僅限於兒童,並必須徵得其法定監護人簽署的知情同意書;當兒童實際上能做出同意參加研究的決定時,還必須徵得其本人同意。
(五) 在緊急情況下,無法事先取得本人或其合法代表的知情同意
書,例如所患的疾病缺乏已被証實有效的治療方法,而試驗
5
(六) 用藥品有望挽救生命,可考慮選擇他們作為受試者,但需要在試驗方案和有關文件中清楚說明接受這些受試者的方法,並事先取得倫理委員會批准在此緊急情況下使用。在給予試驗用藥品之後,研究者須盡快通知受試者或其合法代表有關試驗的事,並得到他們的同意繼續參與試驗。同時,研究者須向倫理委員會報告有關情況。 如發現涉及試驗用藥品的重要新資料,則必須將知情同意書
作書面修改,及在送交倫理委員會批准後,再次取得受試者
同意。
第四章 試驗方案
第二十三條 臨床試驗開始前制定試驗方案,該方案應由研究者與申辦者共同協
定並簽署,經倫理委員會審批後實施。
第二十四條 臨床試驗方案應包括以下內容:
(一) 試驗題目、方案的鑒別號和日期。任何修改應有修改編號
及日期。
(二) 試驗目的,試驗的背景,非臨床研究中有臨床意義的發現
和與該試驗有關的臨床試驗結果、已知對人體的潛在危害
與受益,及試驗藥品存在人種差異的可能。
(三) 申辦者的名稱和地址,試驗單位的地址及電話號碼,研究
者的姓名、資格和地址,申辦者方面的醫學專家的姓名、
資格和地址,臨床實驗室和其他醫學和/或技術部門的名稱
及地址。
(四) 將會按照方案、《中成藥藥品臨床試驗質量管理規範》及其
他相關的法例要求進行試驗的陳述。
(五) 試驗設計的類型、隨機分組方法及設盲的水平。
(六) 受試者的入選標準、排除標準和剔除標準,選擇受試者的
步驟,受試者分配的方法。
(七) 根據統計學原理計算要達到試驗預期目的所需的病例數。
(八) 給予受試者的治療,包括試驗用藥的名稱、劑型、劑量、
給藥途徑、給藥方法、給藥次數、療程和有關合併用藥/
治療的規定,以及對包裝和標籤的說明。
(九) 擬進行的臨床及實驗室檢查項目、其測定次數和在技術可
行的情況下,擬進行的藥代動力學分析等。
(十) 試驗用藥的登記與使用紀錄、遞送、分發方式及儲藏條件
的制度。
(十一) 臨床觀察、隨訪步驟和保證受試者依從性的措施。
(十二) 中止臨床試驗的標準,結束臨床試驗的規定。
6
(十三) 療效評定標準,包括評定參數的方法、觀察時間、記錄與
分析。
(十四) 安全性評定標準,包括評定參數的方法、觀察時間、記錄
與分析。
(十五) 預期受試者參與試驗的時間。
(十六) 試驗期間要測量的主要參數和次要參數。
(十七) 受試者的識別編碼、隨機數字表及病例報告表的保存手續。
(十八) 不良事件的記錄要求和嚴重不良事件的報告方法,處理措
施,隨訪的方式、時間和轉歸。
(十九) 試驗用藥編碼的建立和保存,揭盲方法和緊急情況下破盲
的規定。
(二十) 統計分析計劃,統計分析數據集的定義選擇及計劃進行中
期分析的時間安排。統計分析所採用的顯著性水平,以及
處理缺失數據、未用數據及不合邏輯數據的程序。
(二十一) 數據管理和數據可溯性的規定。
(二十二) 研究者應當提供原數據/文件予試驗有關的監查、稽查、倫
理委員會及有關的藥品監督管理部門以進行視察。
(二十三) 臨床試驗的質量控制與質量保證。
(二十四) 試驗相關的倫理學。
(二十五) 臨床試驗預期的進度和完成日期。
(二十六) 試驗結束後的隨訪和醫療措施。
(二十七) 各方承擔的職責及其他有關規定。
(二十八) 參考文獻。
第二十五條 臨床試驗中,若確有需要,可以按規定程序對試驗方案作出修正。
第五章 研究者的職責
第二十六條 負責臨床試驗的研究者應具備下列條件:
(一) 在合法的醫療機構中具有行醫的資格。
(二) 具有試驗方案中所要求的專業知識和經驗。
(三) 對臨床試驗方法具有豐富經驗或者得到本單位有經驗的研
究者在學術上的指導。
(四) 熟悉申辦者所提供的與臨床試驗有關的資料與文獻。
(五) 有權支配參與該項試驗的人員和設備。
(六) 熟悉《中成藥藥品臨床試驗質量管理規範》,遵守有關法
律、法規和道德規範。
第二十七條 研究者必須詳細閱讀和了解試驗方案的內容,並嚴格按照申辦者同
意及已獲得倫理委員會批准(如有必要,有關的藥品監督管理部門同
意)的方案執行。研究者和申辦者應當在方案上或另立的合同上簽
7
署,確認同意方案。
第二十八條 研究者應了解並熟悉試驗用藥的性質、作用、療效及安全性(包括該
藥品臨床前研究的有關資料),同時也應掌握臨床試驗進行期間發現
的所有與該藥品有關的新信息。如該等新信息與受試者的知情同意
有關的話,研究者應修改書面同意書和其他提供予受試者的書面資
料。修改後的知情同意書和其他提供予受試者的書面資料,都應在
使用之前獲得倫理委員會的批准。
第二十九條 在試驗開始之前,研究者應取得倫理委員會的批准意見。
第三十條 研究者應向倫理委員會提交研究者手冊的當前文本,以作為研究者
向倫理委員會書面申請的一部分。
第三十一條 研究者必須在有良好醫療設施、實驗室設備、人員配備的醫療機構
進行臨床試驗,該機構應具備處理緊急情況的設施,以確保試驗期
間受試者的安全及試驗能正確地進行。實驗室檢查結果必須正確可
靠。
第三十二條 研究者應獲得所在醫院或主管單位的同意,保證有充分的時間在方
案規定的期限內負責和完成臨床試驗。研究者須向參加臨床試驗的
所有工作人員說明有關試驗的資料、規定和職責,確保有足夠數量
並符合試驗方案的受試者進入臨床試驗。
第三十三條 研究者應向受試者說明經倫理委員會同意的有關試驗的詳細情
況,並取得知情同意書。研究者或試驗職員不應強迫或不正當地影
響一個受試者參加或繼續參加試驗。
第三十四條 研究者或由研究者指定的人,應當向每個受試者解釋試驗用藥的正
確用法,並每隔一段時間檢查受試者是否遵從指示使用。
第三十五條 研究者負責作出與臨床試驗相關的醫療決定,保證受試者在試驗期
間出現不良事件時得到適當的治療。
第三十六條 研究者有義務採取必要的措施以保障受試者的安全,並記錄在案。
在臨床試驗過程中如發生嚴重不良事件,研究者應立即安排對受試
者採取適當的治療措施,同時立即報告有關的藥品監督管理部門、
申辦者和倫理委員會,並在報告上簽署及註明日期。
第三十七條 研究者應當遵循試驗的隨機化程序(如有),並應保證依照方案打開
隨機編碼。如果試驗採用盲法,研究者應當保證立即記錄並向申辦
者解釋任何試驗用藥的提前破盲。
第三十八條 研究者應保證將數據真實、準確、完整、及時、合法地載入病歷和
病例報告表。
第三十九條 研究者應接受申辦者派遣的監查員或稽查員的監查和稽查,及有關
的藥品監督管理部門的稽查和視察,以確保臨床試驗的質量。
第四十條 研究者應與申辦者商定有關臨床試驗的費用,並在合同中寫明。 第四十一條 研究者應當每年一次,或應倫理委員會要求的頻率向倫理委員會提 8
交書面的試驗情況摘要。臨床試驗完成後,研究者必須寫出總結報
告,簽署並註明日期後送交申辦者。
第四十二條 研究者中止一項臨床試驗必須通知受試者、申辦者、倫理委員會和
有關的藥品監督管理部門,並闡明理由。
第六章 申辦者的職責
第四十三條 申辦者負責發起、申請、組織和監查一項臨床試驗,並提供試驗經
費。申辦者按相關的法例要求規定,向有關的藥品監督管理部門遞
交臨床試驗的申請,也可委託合同研究組織執行臨床試驗中的某些
工作和任務,但有關試驗數據的質量及完整性的責任最終在於申辦
者。合同研究組織應建立質量保證及質量管理系統。轉移給合同研
究組織或由合同研究組織承擔的任何與臨床試驗有關的職責,應當
有書面說明。沒有明確轉移給合同研究組織或沒有明確寫明由合同
研究組織承擔的職責仍然由申辦者承擔。在本指導原則中涉及申辦
者的一切也適用於一個合同研究組織。
第四十四條 申辦者應選擇有認可資格的臨床試驗機構和研究者以保證試驗的
完成。
第四十五條 申辦者應當指定有合適資格的醫學人員,以便能迅速地對試驗有關
的問題提出建議。
第四十六條 申辦者可考慮設立一個獨立的數據監查委員會(IDMC),定期評價臨
床試驗的進展。
第四十七條 申辦者負責提供最新的研究者手冊,其內容包括試驗用藥品的化
學、藥學、毒理學、藥理學和臨床的(包括以前的和正在進行的試驗)
資料和數據。
第四十八條 申辦者應提供足夠的時間讓研究者審議方案及其他有關的資料。 第四十九條 申辦者在獲得有關的藥品監督管理部門批准並取得倫理委員會批
准後,可以開始按方案組織臨床試驗。
第五十條 申辦者、研究者共同設計臨床試驗方案,方案應述明在實施、數據
管理、統計分析、結果報告、發表論文方式等方面的職責及分工。
雙方應簽署已獲同意的試驗方案及合同。
第五十一條 計劃試驗時,申辦者應當保證有足夠的非臨床研究和/或臨床研究的
安全性及有效性數據,以支持試驗中所採用的給藥途徑、劑量及持
續時間。
第五十二條 申辦者應得到研究者在以下方面的同意:
(一) 按照《中成藥藥品臨床試驗質量管理規範》、相關的法例要求
和經申辦者同意、倫理委員會批准的試驗方案實施臨床試驗
(二) 遵循數據的記錄/報告程序
(三)允許監查、稽查和視察
9
第五十三條 申辦者須確保會按時向研究者提供具有易於識別、正確編碼並貼有
特殊標籤的試驗用藥品、標準品、對照藥品或安慰劑,並保證質量
合格。試驗用藥應按試驗方案的需要進行適當包裝、保存。申辦者
應建立試驗用藥的管理制度和記錄系統,包括運輸、接收、分發、
銷毀、回收及處理未使用的試驗用藥的程序及紀錄。申辦者應決定
試驗用藥的儲存溫度、儲存條件及儲存時間等。申辦者並應確保研
究者遵循有關的書面操作程序處理試驗用藥。
第五十四條 申辦者應採取措施以保證試驗用藥在整個使用期內的穩定性,並保
存足夠數量的試驗用藥,以便在有需要時可以再確認其產品規格。
同時申辦者應保存試驗用藥的批樣分析和特性紀錄。
第五十五條 申辦者應持續地就試驗用藥品的安全性進行評價。
第五十六條 申辦者應確保試驗得到適當的監查,通常需要在試驗前、試驗期間
和試驗後到試驗單位進行現場監查。申辦者任命合資格並為研究者
所接受的監查員,以監查臨床試驗的進行,並寫出監查報告。
第五十七條 申辦者應確保在方案或其他的書面協議中說明研究者須允許與試
驗有關的監查員、稽查員、倫理委員會或管理部門直接使用原數據
/文件。
第五十八條 申辦者應建立對臨床試驗的質量控制和質量保證系統,並可組織對
臨床試驗的稽查以保證質量。
第五十九條 申辦者應與研究者迅速研究所發生的嚴重不良事件,採取必要的措
施以保證受試者的安全和權益,並立即向有關的藥品監督管理部門
及倫理委員會報告,同時向涉及同一藥品的臨床試驗的其他研究者
通報。
第六十條 申辦者中止一項臨床試驗前,須通知研究者、倫理委員會和有關的
藥品監督管理部門,並述明理由。
第六十一條 申辦者負責向有關的藥品監督管理部門遞交試驗的總結報告。
第六十二條 申辦者應對參加臨床試驗的受試者提供保險,對於發生與試驗相關
的損害或死亡的受試者承擔治療的費用及相應的經濟補償。申辦者
應向研究者提供法律上與經濟上的擔保,但由醫療事故導致者除
外。
第六十三條 試驗的財務安排應當列入申辦者和研究者之間的協議中。
第六十四條 研究者不遵從已批准的方案或相關法例要求進行臨床試驗時,申辦
者應指出以求糾正,如情況嚴重或持續不改,則應終止研究者參加
臨床試驗並向藥品監督管理部門報告。
第七章 監查員的職責
第六十五條 監查的目的是為了保證臨床試驗中受試者的權益受到保障,試驗紀
錄與報告的數據準確、完整無誤,保證試驗遵循已批准的方案、《中
10
成藥藥品臨床試驗質量管理規範》和相關的法例要求進行。
第六十六條 監查員是申辦者與研究者之間的主要聯繫人。監查員應由申辦者指
定。其人數及訪視的次數取決於臨床試驗的複雜程度和參與試驗的
醫療機構的數目。監查員應有適當的醫學、藥學或相關專業學歷,
並經過必要的訓練,熟悉藥品管理有關法規,熟悉有關試驗用藥品
的臨床前和臨床方面的信息以及臨床試驗方案及其相關的文件。
第六十七條 監查員應遵循標準操作規程,督促臨床試驗的進行,以保證臨床試
驗按方案執行。具體內容包括:
(一) 在試驗前確認試驗機構已具有適當的條件,包括人員配備
與培訓情況,實驗室設備齊全、運轉良好,具備各種與試
驗有關的檢查條件,參與研究人員熟悉試驗方案中的要求。
(二) 在試驗過程中監查研究者對試驗方案的執行情況,確定研
究者在試驗之前已獲得研究者手冊,確定在試驗前已取得
所有受試者的知情同意書,了解受試者的入選率及試驗的
進展狀況,確認入選的受試者合格。
(三) 確認所有數據的紀錄與報告正確完整,所有病例報告表填
寫正確,並與原始資料一致。所有錯誤或遺漏均已改正或
註明,並經研究者簽名及註明日期。任何的劑量改變、治
療變更、合併用藥、間發疾病、失訪、檢查遺漏等均應確
認並記錄。核實入選受試者的退出與失訪已在病例報告表
中予以說明。
(四) 確認所有不良事件均已記錄在案,而嚴重不良事件亦在規
定時間內作出報告並記錄在案。
(五) 核實試驗用藥是否按照相關的法例要求進行供應、儲藏、
分發、回收,並做相應的紀錄。
(六) 協助研究者進行必要的通知、申請事宜,並向申辦者報告
試驗數據和結果。
(七) 應清楚如實記錄研究者未能做到的隨訪、未進行的試驗和
未做的檢查,以及是否對錯誤、遺漏作出糾正。
(八) 確定研究者是否有保存必需文件(見附錄三)。
(九) 核實研究者及其試驗職員是否按照方案及其他申辦者與研
究者的書面協議執行指定的職務,及沒有將這些職務委派
給未經授權的人。
(十) 每次訪視後作一書面報告遞送申辦者,報告應述明監查日
期、時間、監查員姓名、監查的發現、曾接觸過的研究者
的姓名等。
紀錄與報告
11 第八章
第六十八條 病歷是臨床試驗的原始文件,應完整保存。病例報告表中的數據來
自原始文件並與原始文件一致,試驗中的任何觀察及檢查結果均應
及時、準確、完整、規範、真實地記錄於病歷和正確地填寫至病例報告表中,不得隨意更改,確因填寫錯誤而作任何更正時,應保持
原紀錄清晰可辨,由更正者簽署並註明日期。
第六十九條 臨床試驗中各種實驗室數據均應記錄或將原始報告複印件黏貼在
病例報告表上,在正常範圍內的數據也應具體記錄。對顯著偏離或
在臨床可接受範圍以外的數據須加以核實。檢測項目必須註明所採
用的計量單位。
第七十條 為保護受試者私隱,病例報告表上不應出現受試者的姓名。研究者
應有受試者的識別代碼和確認紀錄。
第七十一條 臨床試驗總結報告內容應與試驗方案要求一致,包括:
(一) 隨機進入各組的實際病例數,退出試驗和剔除的病例及其
理由;
(二) 不同組間的基線特徵比較,以確定可比性;
(三) 對所有療效評價指標進行統計分析和臨床意義分析。統計
結果的解釋應着重考慮其臨床意義;
(四) 安全性評價應有臨床不良事件和實驗室指標合理的統計分
析,對嚴重不良事件應詳細描述和評價;
(五) 多中心試驗中評價療效時,應考慮各中心之間存在的差異
及其影響;
(六) 對試驗用藥的療效和安全性以及風險和受益之間的關係作
出簡要概述和討論。
第七十二條 臨床試驗中的必需文件均須按規定保存(附錄3)及管理。研究者
應保存臨床試驗必需文件至臨床試驗終止後五年。申辦者應保存臨
床試驗資料至試驗藥品被批准上市後五年。
第九章 數據管理與統計分析
第七十三條 數據管理的目的在於把試驗數據迅速、完整、無誤地納入報告,所
有涉及數據管理的步驟均須記錄在案,以便對數據質量及試驗實施
進行檢查。用適當的程序保證數據庫的保密性,應具有計算機數據
庫的維護和支持程序。
第七十四條 臨床試驗中受試者分配必須按試驗設計確定的隨機分配方案進
行,每名受試者的處理分組編碼應作為盲底由申辦者和研究者分別
保存。設盲試驗應在方案中規定揭盲的條件和執行揭盲的程序,並
配有相應處理編碼的應急信件。在緊急情況下,允許對個別受試者
緊急破盲而了解其所接受的治療,但必須在病例報告表上述明理
由。
12
第七十五條 臨床試驗資料的統計分析過程及其結果的表達必須採用規範的統
計學方法。臨床試驗各階段均需有生物統計學專業人員參與。臨床
試驗方案中需有統計分析計劃,並在正式統計分析前加以確認和細
化。若需作中期分析,應說明理由及操作規程。對治療作用的評價
應將可信區間與假設檢驗的結果一併考慮。所選用統計分析數據集
需加以說明。對於遺漏、未用或多餘的資料須加以說明,臨床試驗
的統計報告必須與臨床試驗總結報告相符。
第七十六條 應用電子數據處理和/或遙控電子試驗數據系統時,申辦者應當:
(一) 確保並證明電子數據處理系統符合申辦者所設定的關於完整
性、準確性、可靠性和一致預期的性能(如數據確認)的要求;
(二) 為使用這個系統保存一份標準操作規程;
(三) 保證系統的設計允許數據修改按如下方式進行:數據的改變會
被記錄而不刪除原本的數據(即保留稽查軌跡、數據軌跡和編
輯軌跡);
(四) 有一個防止未經授權接觸數據的安全系統;
(五) 有一份被授權修改數據的人員名單;
(六) 保存足夠的數據備份;
(七) 如採用盲法,建立保護盲法的措施(例如:在數據輸入和處理
期間維持盲法。
第七十七條 如在處理過程中曾經作過數據的轉換,應當隨時都可將原數據與已
處理的數據作比較。
第十章 試驗用藥的管理
第七十八條 在臨床試驗中使用的試驗用藥,未經批准,不得在市場上經銷。 第七十九條 申辦者負責對臨床試驗用藥作適當的包裝與標籤,並標明為臨床試
驗專用。在雙盲臨床試驗中,試驗用藥品與對照藥品或安慰劑在各
項特徵上均應一致,包括外形、包裝、標籤等等。但在醫學緊急情
況下,應可允許迅速鑒別藥品而不會有不被察覺的破盲。
第八十條 試驗用藥的使用紀錄應包括數量、裝運、遞送、接受、分配、應用
後剩餘藥品的回收與銷毀等方面的信息。
第八十一條 試驗用藥的使用由研究者負責,研究者必須保證所有試驗用藥僅用
於該臨床試驗的受試者,其劑量與用法應遵照試驗方案,而剩餘的
試驗用藥應退回申辦者。上述過程須由專人負責並記錄在案,試驗
用藥亦須有專人管理。研究者不得把試驗用藥轉交任何非臨床試驗
參加者。
第八十二條 試驗用藥的供給、使用、儲藏及剩餘藥品的處理過程應接受相關人
員的檢查。
第十一章 質量保證
13
第八十三條 為保證試驗的進行以及數據的產生、記錄及報告都符合《中成藥藥
品臨床試驗質量管理規範》,應建立有計劃及有系統的質量保證的
活動。申辦者及研究者均應履行各自職責,並嚴格遵循臨床試驗方
案, 採用標準操作規程,以保證臨床試驗的質量控制和質量保證系
統的實施。
第八十四條 臨床試驗中所有觀察結果和發現都加以核實,在數據處理的每一階
段必須進行質量控制,以保證數據完整、準確、真實、可靠。
第八十五條 藥品監督管理部門、申辦者可委託稽查人員對臨床試驗進行系統性
檢查,以判定試驗的執行是否與試驗方案相符,報告的數據是否與
各臨床參加單位的紀錄一致,即病例報告表記錄的數據是否與病歷
或其他原始紀錄相同。稽查應由不直接涉及該臨床試驗的人員執
行。稽查人員應對於臨床試驗中的造假、欺詐行為作出報告。
第八十六條 作為質量保證的一部份,申辦者進行稽查時應考慮:
(一) 獨立而又與一般常規的監查或質量控制分開的稽查,目的是評
價試驗的實施和對試驗方案、標準操作規程及《中成藥藥品臨
床試驗管理規範》等等的依從性。
(二) 申辦者應指定一個獨立於臨床試驗的人實施稽查,而該人應經
過培訓並有相關經驗。
(三) 申辦者應確保稽查的程序,包括稽查的對象、方法、頻率以及
內容都必須按照書面程序進行。
(四) 申辦者所訂定的稽查計劃及程序應根據試驗提交予有關的藥
品監督管理部門的重要性、受試者的數目、試驗的類型及其複
雜程度、受試者的風險水平等而定。
(五) 就稽查員的觀察和發現應作記錄。
(六) 為保持稽查員的獨立性及價值,有關的藥品監督管理部門不應
定期地要求稽查報告。但當有嚴重不依照《中成藥藥品臨床試
驗質量管理規範》時,或在法律訴訟期間,有關的藥品監督管
理部門可個別要求稽查報告。
(七) 在有關的法律或法規要求下,申辦者應當提供稽查證書。
第八十七條 有關的藥品監督管理部門應對研究者與申辦者在實施試驗中各自
的任務與執行狀況進行視察。參加臨床試驗的醫療機構和實驗室所
有資料(包括病歷)及文件均應準備接受有關的藥品監督管理部門
的視察。
第十二章 多中心試驗
第八十八條 多中心試驗是由多位研究者按同一試驗方案在不同地點和單位進
行的臨床試驗。多中心試驗由一位主要研究者總負責,並作為臨床
試驗各中心間的協調研究者。
14
第八十九條 多中心試驗的計劃和組織實施要考慮以下各點:
(一) 試驗方案及其附件由各中心的主要研究者與申辦者共同討
論認定,倫理委員會批准後執行。
(二) 應在臨床試驗開始時及進行的中期時組織研究者會議。
(三) 各中心臨床試驗樣本大小及中心間的分配應符合統計分析
的要求。
(四) 保證在不同中心以相同程序管理試驗用藥,包括分發和儲
藏。
(五) 根據同一試驗方案培訓參加該試驗的研究者。
(六) 建立標準化的評價方法,試驗中所採用的實驗室和臨床評
價方法均應有統一的質量控制,實驗室檢查也可由中心實
驗室進行。
(七) 數據資料應集中管理與分析,應建立數據傳遞、管理、核
查與查詢程序。
(八) 保證各試驗中心研究者遵從試驗方案,包括在違背方案時
終止其參加試驗。
第九十條 多中心試驗應根據參加試驗的中心數目和試驗的要求,以及對試驗
用藥品的了解程度建立管理系統,協調研究者負責整個試驗的實
施。
第十三章 研究者手冊
第九十一條 研究者手冊列明了有關試驗用藥品的臨床和非臨床資料。手冊旨在
向研究者及其他參與試驗的人員提供資料,幫助他們了解方案中的
關鍵特徵及其基本原理,並遵循這些關鍵特徵,如劑量、給藥次數
/相距的時間、給藥方法和安全性監查程序。研究者手冊也為受試者
在臨床試驗過程中的臨床管理提供指示。有關的資料應以簡單、易
明、客觀、公平、非宣傳性的形式,使醫生或註冊中醫師或有可能
成為研究者的人明白手冊的內容,就建議中的試驗的合理性作出他
們中立的風險/利益評估。因此,一般來說合資格的醫學人士應參與
研究者手冊的編寫,而手冊的內容必須得到提供資料的學科認可。
第九十二條 本指引描述了研究者手冊應當包含資料的最低要求,並為其編排作
出了建議。預料所得資料的類型和範圍,將隨試驗用藥品的開展階
段而有所變化。當試驗用藥品上市,而其藥理已廣為醫學人員了解
後,就可能不再需要一本詳細的研究者手冊。
第九十三條 若有關的藥品監督管理部門許可,產品資料手冊、包裝說明或標籤
都可以作為合適的代替品,惟該等物品須載有試驗用藥各方面的最
新、全面而詳盡的資料,這點對研究者殊為重要。
第九十四條 如果正在研究一個已上市產品的新用途(即一個新主治病證),應當 15
特別準備一份關於該新用途的研究者手冊。
第九十五條 研究者手冊至少應每年審評一次,並在有需要時按申辦者的書面程
序修改。根據新藥的發展階段新資料的衍生,手冊可能需要更頻繁
的修改。但是,依照《中成藥藥品臨床試驗質量管理規範》要求,
有關的新資料可能很重要,在考慮將其列入研究者手冊之前,須事
先通知研究者、倫理委員會和/或有關的藥品監督管理部門。
第九十六條 一般而言,申辦者負責保證向研究者提供最新的研究者手冊,研究
者有責任將最新的研究者手冊提供予負責的倫理委員會。在由研究
者申辦試驗時,申辦者(研究者)應當決定手冊是否可從製造商處得
到。
第九十七條 如果試驗用藥是由申辦者(研究者)提供的話,他須向試驗有關的人
員提供所需資料。如果不可能準備到一份正式的研究者手冊,申辦
者(研究者)也應增加試驗方案中的背景資料,最少應包含本指引所
載必需的最新資料。
第九十八條 一般資料:
研究者手冊應當包括:
(一) 封面:封面應當提供申辦者姓名,每一個試驗用藥的鑒別
(即研究編號、藥品名或已批准的通用名、申辦者擬定的商
品名),以及發佈日期。此外還建議列出版本號碼的參考索
引,以及該版本替換和被批准的日期。示例見附頁2。
(二) 保密性陳述:申辦者可能希望包括一段陳述,指示研究者/
收件人將研究者手冊作機密文件處理,並僅供研究人員小
組和倫理委員會使用。
第九十九條 研究者手冊的內容
研究者手冊應當包括下列章節,應根據需要於適當的章節附上參考
文獻:
(一) 目錄:附頁3為目錄的例子
(二) 摘要:應有一個簡短的摘要(最好不超過2頁),重點是與
試驗用藥品發展階段有關的、有意義的臨床和非臨床資料。
(三) 前言:應當有一個簡短的前言,說明試驗用藥品的藥品名
(批准時的通用名和商品名)、試驗用藥品正在進行研究的
基本原理、預期的主治病證。最後,前言應當提供評價試
驗用藥品的一般方法。
(四) 理化及藥學特性和處方:應提供有關於試驗用藥品的理化
和藥學特性的簡短摘要。在試驗過程中允許採取合宜的安
全措施,如果臨床上相關,則應提供所用配方包括賦形劑
的描述,並應提出配方理由,也應作出製劑儲存和處理的
說明。
16
(五) 非臨床研究:
前言: 應當以摘要形式提供所有非臨床的藥理學、毒理學研究的
有關結果。如技術可行,再加上藥代動力學和試驗用藥的
代謝研究的結果。摘要應當說明所採用的方法學、結果,
以及這些發現與研究中的主治病證的關係,和對人類可能
產生的不利及非預期中的影響。
如可行的話,必須提供以下資料:
z 試驗動物種屬
z 每組動物的數目和性別
z 劑量單位(如:毫克/千克)
z 給藥相距的時間
z 給藥途徑
z 給藥持續時間
z 有關藥品在全身分布的資料
z 給藥後隨訪持續的時間
z 結果,包括下列方面:
- 藥理作用或毒性作用的性質和頻率
- 藥理作用或毒性作用的嚴重性或強度
- 開始作用時間
- 作用的可逆性
- 作用持續的時間
- 劑量反應
在可能的情況下,應採用表格/列表以增強陳述的清晰度。
隨後的章節應當討論研究的最重要發現,包括觀察所得的
劑量反應關係,與人類的相關性,以及在人體上研究的各
個方面。如果合適,在同一動物種屬的有效劑量和非毒性
劑量的發現,應當作出比較(即應討論治療指數)。應當說
明這一資料與所提議的人用劑量的相關性。在可能情況
下,這些比較應根據血/組織水平而非毫克/千克進行。
1. 非臨床藥理學
應當包括試驗用藥品的藥理學研究的摘要,如有可能還
應包括藥品在動物的重要代謝研究摘要。這樣一個摘要
應當涵蓋潛在治療活動(如有效模型、受體結合和特異
性評估研究)以及評價安全性的研究(如不同於治療作用
的評價藥理學作用的專門研究) 。
2. 動物的藥代動力學和藥物代謝(如技術上可行)
應提供試驗用藥品在所研究種屬動物中的藥代動力
17
(六) 學、生物轉化以及處置的摘要。對發現的討論應當說明試驗用藥及其代謝物的吸收及其局部、全身的生物利用度,以及與動物藥理學和毒理學的關係。 3. 毒理學 在不同動物種屬中進行相關研究所發現的毒理作用摘要,應按以下標題描述: -單劑量給藥 -重覆給藥 -致癌性(如適用) -特殊毒理研究(如刺激性和致敏性) (如適用) -生殖毒性(如適用) -遺傳毒性(致突變性) (如適用) 對人類的作用 前言
應提供有關試驗用藥品對人類產生的已知作用的充分討論,包括關於藥效學、劑量反應、安全性、有效性和其他藥理學領域(如技術上可行,還應包括藥代動力學及代謝)。如有可能,應提供每一個已經完成的臨床試驗的摘要。還應提供試驗用藥品在臨床試驗以外的用途的結果,如上市期間的經驗。
1. 試驗用藥品在人體的藥代動力學和代謝(如技術上可行)
應寫出試驗用藥品的藥代動力學資料摘要,包括以下方面:
-藥代動力學(包括代謝和吸收、血漿蛋白結合、分布
及排泄)
-試驗用藥品的標準劑型的生物利用度(絕對和相對生
物利用度)
-人群亞組(如性別、年齡和臟器功能損害)
-相互作用(如藥物之間的相互作用和藥物與食物的相
互作用)
-其他藥代動力學數據(如在臨床試驗期間完成的群體
研究結果)
2. 安全性和有效性
-應提供以往在人體試驗(健康志願者和/或病人)中所獲得關於試驗用藥品(包括代謝物)的安全性、藥效學、有效性和劑量反應資料的摘要,並應討論這些資料的含義。當已有一定數目的臨床試驗完成後,使用從不同試驗中所得到關於人群亞組主治病證的安全性及成
18
(七) 效性的摘要,可為臨床試驗提供清晰的資料。從所有臨床試驗(包括那些作成效性考核的)得到的藥品不良反應資料,可以用表格形式表達。至於在主治病證或亞組之間的藥品不良反應類型/發生率的重要差異,則應進行討論。 根據試驗用藥品及其相關產品先前的使用經驗,研究者應提供可能預見的危害及不良藥品反應的描述。此外,有關試驗用藥品的注意事項及其他須採取的特別監察措施亦須清楚說明。 3. 銷售經驗 研究者手冊應識別試驗用藥品已經上市或已獲批准的國家。從上市使用中得到的任何重要資料應當作簡要陳述(如處方、劑量、給藥途徑和藥品不良反應)。研究者手冊也應識別試驗用藥品還未獲批准/註冊上市或退出上市/註冊的所有國家。 數據和研究人員指南摘要
本節應提供一個非臨床數據的全面討論,只要可能,應對試驗用藥品不同方面的各種來源的資料作一摘要。研究者如此便可得到現有數據最具資料性的解釋,以及這些資料對於將來臨床試驗意義的評價。
如可行應對有關產品已發表的報告進行討論。這有助研究者預測藥品不良反應或臨床試驗中的其他問題。
這一節的總目的是讓研究者清楚明白可能出現的風險和不良反應,以及臨床試驗中可能需要的特殊檢查、觀察資料和防範措施。這種了解應以研究該藥品的理化、藥學、藥理、毒理和臨床資料為基礎。根據先前人類的經驗和試驗用藥品的藥理學,就可能出現的服藥過量和藥品不良反應,向研究者提供識別和處理的指引。
19
附頁1 臨床試驗的人員架構圖

- 不直接涉及試驗的人員所進行的一種系統性檢查,以判斷
試驗的實施、數據的記錄及分 析是否與試驗方案、《中成藥藥 品臨床試驗質量管理規範》與法規要求相符

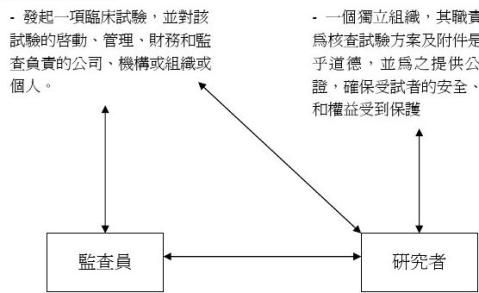
- 由申辦者任命並對申辦者負責的具備相關知識的人員,其任務是監查和報告試驗的進行情況和核實數據 - 實施臨床試驗並對臨床試驗的質量及受試者安全和權益的負責者
20
附頁2
研究者手冊封面(示例) 申辦者名稱
藥品:
1. 研究代號:
2. 藥品名,通用名(若已批准)
3. 商品名(若符合申辦者意願)
研究者手冊
版本編號:
發佈日期:
被替代的版本編號:(如適用) 日期:
21
附頁3
研究者手冊目錄(舉例)
- 保密性陳述(如有)
- 簽署頁(如有)
1. 目錄
2. 概要
3. 引言
4. 藥學特性和處方
5. 非臨床研究
6. 人體內作用
7. 數據概要和研究者指引
注意: 參考資料 1. 公開發表文件
2. 報告 參考資料應在章節末列出
附錄(如有)
22
世界醫學大會赫爾辛基宣言
涉及人類受試者醫學研究的倫理學原則
通過 : 第18屆世界醫學大會, 赫爾辛基, 芬蘭, 19xx年6月
修訂 : 第29屆世界醫學大會, 東京, 日本, 19xx年10月
第35屆世界醫學大會, 威尼斯, 意大利, 19xx年10月
第41屆世界醫學大會, 香港, 19xx年9月
第48屆世界醫學大會, Somerset West, 南非共和國, 19xx年10月 第52屆世界醫學大會, 愛丁堡, 蘇格蘭, 20xx年10月
一. 引言
(一) 世界醫學大會起草的赫爾辛基宣言,是人體醫學研究倫理準則的聲
明,用以指導醫生及其他參與者進行人體醫學研究。人體醫學研究包括對人體本身和相關數據或資料的研究。
(二) 促進和保護人類健康是醫生的職責。醫生的知識和道德是為了履行
這一職責。
(三) 世界醫學大會的日內瓦宣言用“病人的健康必須是我們首先考慮
的事”。這樣的語言對醫生加以約束。醫學倫理的國際準則宣告:“只有在符合病人的利益時,醫生才可提供可能對病人的生理及心理產生不利影響的醫療措施”。
(四) 醫學的進步以研究作為基礎,這些研究在一定程度上最終有賴於以
人作為受試者的試驗。
(五) 在人體醫學研究中,對受試者健康的考慮應優先於科學和社會的興
趣。
(六) 人體醫學研究的主要目的是改進預防、診斷和治療方法,提高對疾
病病因學和發病機理的認識。即使是已被證實了的最好的預防、診斷和治療方法都應不斷的通過研究來檢驗其有效性,效率,可行性和質量。
(七) 在目前的醫學實踐和醫學研究中,大多數的預防、診斷和治療都包
含有風險和負擔。
(八) 醫學研究應遵從倫理標準,對所有的人加以尊重並保護他們的健康
和權益。有些受試人群是弱勢群體需加以特別保護。必須認清經濟和醫療上處於不利地位的人的特殊需要。要特別關注那些不能做出知情同意或拒絕知情同意的受試者、可能在脇迫下才做出知情同意的受試者、從研究中本人得不到受益的受試者及同時接受治療受試者。
23
(九) 研究者必須知道所在國關於人體研究方面的倫理、法律和法規的要
求,並且要符合國際的要求。任何國家的倫理、法律或法規都不允許減少或取消本宣言中對受試者所規定的保護。
二. 醫學研究的基本原則
(十) 在醫學研究中,保護受試者的生命和健康,維護他們的隱私和尊
嚴是醫生的職責。
(十一) 人體醫學研究必須遵從普遍接受的科學原則,並基於對科學文獻
和相關資料的全面了解及充分的實驗室試驗和動物試驗(如有必
要)。
(十二) 必須適當謹慎地實施可能影響環境的研究,並要尊重用於研究的
實驗動物的權利。
(十三) 每項人體試驗的設計與實施均應在試驗方案中明確說明,並應將
試驗方案提交給倫理審批委員會進行審核、評論、指導,適當情
況下,進行審核批准。該倫理委員會必須獨立於研究者和申辦者,並且不受任何其他方面的影響。該倫理委員會應遵從試驗所在國
的法律和制度。委員會有權監督進行中的試驗。研究人員有責任
向委員會提交監查資料,尤其是所有的嚴重不良事件的資料。研
究人員還應向委員會提交其他資料以備審批,包括有關資金、申
辦者、研究機構以及其它對受試者潛在的利益衝突或鼓勵的資料。 (十四) 研究方案必須有關於倫理方面的考慮的說明,並表明該方案符合本
宣言中所陳述的原則。
(十五) 人體醫學研究只能由有專業資格的人員並在臨床醫學専家的指導
監督下進行。必須始終是醫學上有資格的人員對受試者負責,而決
不是由受試者本人負責,即使受試者已經知情同意參加該項研究。 (十六) 每項人體醫學研究開始之前,應首先認真評價受試者或其他人員的
預期風險、負擔與受益比。這並不排除健康受試者參加醫學研究。所有研究設計都應公開可以獲得。
(十七) 醫生只有當確信能夠充分地預見試驗中的風險並能夠較好地處理
的時候才能進行該項人體研究。如果發現風險超過可能的受益或已
經得出陽性的結論和有利的結果時醫生應當停止研究。
(十八) 人體醫學研究只有試驗目的的重要性超過了受試者本身的風險和
負擔時才可進行。這對受試者是健康志願者時尤為重要。
(十九) 醫學研究只有在受試人群能夠從研究的結果中受益時才能進行。 (二十) 受試者必須是自願參加並且對研究項目有充分的了解。
(二十一)必須始終尊重受試者保護自身的權利。盡可能採取措施以尊重受
試者的隱私、病人資料的保密並將對受試者的身體和精神以及人
格的影響減至最小。
24
(二十二)在任何人體研究中都應向每位受試候選者充分地告知研究的目
的、方法、資金來源、可能的利益衝突、研究者所在的研究附屬機構、研究的預期的受益和潛在的風險以及可能出現的不適。應告知受試者有權拒絕參加試驗或在任何時間退出試驗並且不會受到任何報復。當確認受試者理解了這些信息後,醫生應獲得受試者自願給出的知情同意,以書面形式為宜。如果不能得到書面的同意書,則必須正規記錄非書面同意的獲得過程並要有見證。
(二十三)在取得研究項目的知情同意時,應特別注意受試者與醫生是否存
在依賴性關係或可能被迫同意參加。在這種情況下,知情同意的獲得應由充分了解但不參加此研究與並受試者也完全無依賴關係的醫生來進行。
(二十四)對於法律上沒有資格,身體或精神狀況不允許給出知情同意,或
未成年人的研究受試者,研究者必須遵照相關法律,從其法定全權代表處獲得知情同意。只有該研究對促進他們所代表的群體的健康存在必需的意義,或不能在法律上有資格的人群中進行時,這些人才能被納入研究。
(二十五)當無法定資格的受試者,如未成年兒童,實際上能作出參加研究
的決定時,研究者除得到法定授權代表人的同意,還必須徵得本人的同意。
(二十六)有些研究不能從受試者處得到同意,包括委託人或先前的同意,
只有當受試者身體/精神狀況不允許獲得知情同意是這個人群的必要特徵時,這項研究才可進行。應當在試驗方案中闡明致使參加研究的受試者不能作出知情同意的特殊原因,並提交倫理委員會審查和批准。方案中還需說明在繼續的研究中應盡快從受試者本人或法定授權代理人處得到知情同意。
(二十七)作者和出版者都要承擔有倫理責任。在發表研究結果時,研究者
有責任保證結果的準確性。與陽性結果一樣,陰性結果也應發表或以其他方式公之於眾。出版物中應說明資金來源、研究附屬機構和任何可能的利益衝突。與本宣言中公布的原則不符的研究報告不能被接受與發表。
三. 醫學研究與醫療相結合的附加原則
(二十八)醫生可以將醫學研究與醫療措施相結合,但僅限於該研究已被證
實具有潛在的預防、診斷和治療價值的情況下。當醫學研究與醫療措施相結合時,病人作為研究的受試者要有附加條例加以保護。 (二十九)新方法的益處、風險、負擔和有效性應當與現有最佳的預防、診
斷和治療方法作對比。這並不排除在目前沒有有效的預防、診斷和治療方法存在的研究中,使用安慰劑或無治療作為對照。
25
(三十) 在研究結束時,每個入組病人都應當確保得經該研究證實的最有
效的預防、診斷和治療方法。
(三十一)醫生應當充分告知病人其接受的治療中的那一部分與研究有關。
病人拒絕參加研究絶不應該影響該病人與醫生的關係。
(三十二)在對病人的治療中,對於沒有已被證明的預防、診斷和治療方法,
或在使用無效的情況下,若醫生判定一種未經證實或新的預防、
診斷和治療方法有望挽救生命、恢復健康和減輕痛苦,在獲得病
人的知情同意的前題下,應不受限制地應用這種方法。在可能的
情況下,這些方法應被作為研究對象,並有計劃地評價其安全性
和有效性。記錄從所有相關病例中得到的新資料,適當時予以發
表。同時要遵循本宣言的其他相關原則。
26
附錄二
名詞釋義
1. 不良事件 (Adverse Event):病人或臨床試驗受試者接受一種藥品後出
現的不良醫學事件,但並不一定與治療有因果關係。一件不良事件可以
是任何與使用(試驗用)藥品時的不利的及非預期的徵兆、症狀或疾病。
2. 中立的見證人(Impartial Witness):獨立於臨床試驗、不受與試驗有關
人員的不公正影響的個人。如果受試者或其合法代表不能閱讀,他將參
與知情同意過程,並閱讀提供予受試者的知情同意書和其他書面資料。
3. 中成藥藥品臨床試驗質量管理規範(GCP):是臨床試驗設計、實施、執
行、監查、稽查、記錄、分析和報告的標準,它為數據和報告結果的可
信性和準確性提供了保證,並保護受試者的權利、完整性和機密性。
4. 文件(Documentation):描述或記錄試驗的方法、實施和/或結果,影響
試驗的因素,以及採取的措施等的任何形式的記錄(包括但不限於書面、
電子、磁性和光學的記錄,以及掃描、X射線和心電圖)。
5. 未預期的藥品不良反應(Unexpected Adverse Drug Reaction):性質或嚴
重程度與現有的產品資料(例如未批准的試驗用藥品的研究者手冊,或已
經批准藥品的產品資料手冊或說明書)不符的不良反應。
6. 申辦者 (Sponsor):發起一項臨床試驗,並對該試驗的啟動、管理、財
務和監查負責的公司、機構或組織或個人。
7. 申辦者(研究者)(Sponsor-investigator) :單獨或與其他人發起並實施一個
臨床試驗的個人,根據其直接指示將試驗用藥給予受試者使用。申辦者
(研究者)的責任包含申辦者及研究者兩方面。
8. 必需文件(Essential Document):指各自和集合一起允許評價一個研究的
執行情況和所得數據的質量的文件。
9. 多中心試驗(Multicentre Trial):按照一個試驗方案,在一個以上試驗單
位實施,因此由一名以上研究者完成的臨床試驗。
10. 合同(Contract):有關雙方或以上的書面、註明日期及簽署的協議,其
中陳述了關於工作和責任的委託和分派的安排,以及關於財務問題的安
排。試驗方案可以作為合同的基礎。
11. 合同研究組織 (Contract Research Organization, CRO):一種學術性或
商業性的科學機構或個人。申辦者可委託其執行臨床試驗中的某些工作
和任務,此種委託必須作出書面規定。
12. 合法代表(Legally Acceptable Representative):在適用法律下被授權代
表預期的受試者同意參加臨床試驗的個人,或司法人員或其他團體。
13. 批准(倫理委員會)(Approval(in relation to Institutional Review Board)) :
倫理委員會表示贊成的決定,表示對該臨床試驗已經進行審評,並同意
可在倫理委員會、研究機構、《中成藥藥品臨床試驗質量管理規範》或
其他法規的約束下進行該試驗。
27
知情同意 (Informed Consent):指向受試者告知一項試驗的各個方面情況後,受試者自願確認其同意參加該項臨床試驗的過程,須以簽名和註明日期的知情同意書作為文件證明。
15. 知情同意書 (Informed Consent Form):是每位受試者表示自願參加某
一試驗的文件證明。研究者需向受試者說明試驗性質、試驗目的、可能的受益和風險、可供選用的其他治療方法以及符合《赫爾辛基宣言》規定的受試者的權利和義務等,使受試者充分了解後表達其同意。
16. 依從性(關於試驗的)(Compliance(in relation to trials)):遵循與試驗有關
的所有要求、《中成藥藥品臨床試驗質量管理規範 (GCP)》和相關的法例要求。
17. 直接使用(Direct Access):允許閱覽、分析、核對和複製任何對於評價臨
床試驗有重要意義的記錄與報告。允許直接使用的一方(如國內和國外的藥品監督管理部門,申辦者方的監查員和稽查員)應當根據相關的法例要求,採取一切合理的預防措施維護受試者身份和申辦者資料的保密性。 18. 受試者(Subject/Trial Subject):參加一個臨床試驗作為試驗藥品的接受
者或作為對照的個人。
19. 受試者識別編碼(Subject Identification Code):研究者為每一名受試者
指定的獨特識別號碼,以保護受試者的身份並在研究者報告不良事件和/或其他與試驗有關數據時,代替受試者姓名。
20. 協調研究者 (Coordinating Investigator):在多中心臨床試驗中負責協調
參加各中心研究者工作的一名研究者。
21. 研究者 (Investigator):實施臨床試驗並對臨床試驗的質量及受試者安全
和權益的負責者。研究者必須經過資格審查,具有臨床試驗的專業特長、資格和能力。在多中心臨床試驗中,由一名主要研究者對臨床試驗實施的總負責,並作為各試驗中心間的協調人。
22. 研究者手冊(Investigator’s Brochure):是有關試驗用藥品在進行人體
研究時已有的臨床與非臨床研究資料。
23. 原文件(Source Documents):原始文件、數據和記錄(如醫院記錄,臨
床和辦公室圖表,實驗室筆記,備忘錄,受試者日記卡或評價表,藥房發藥記錄,自動儀器的記錄數據,經核證的複印件或抄件,顯微膠片,攝影底片,縮微膠卷或磁介質,X線,受試者檔案,以及保存在藥房、實驗室和參與臨床試驗的醫學技術部門中的記錄。)
24. 病例報告表 (Case Report Form, CRF):指按試驗方案所規定設計的一
種文件,用以記錄每一名受試者在試驗過程中的數據。
25. 倫理委員會 (Ethics Committee):由醫學專業人員、法律專家及非醫務
人員組成的獨立組織,其職責為核查臨床試驗方案及附件是否合乎道德,並為之提供公眾保證,確保受試者的安全、健康和權益受到保護。該委員會的組成和一切活動不應受臨床試驗組織和實施者的干擾或影
2814.
26. 27. 28. 29. 30. 31. 32. 33. 34. 35. 36. 37. 38. 39. 響。 原數據(Source Data):臨床試驗中的臨床發現、觀察或其他活動的原始記錄及其核證副本中的數據,它們對於重建和評價試驗是必要的。原數據應包含在原文件中。 設盲 (Blinding/Masking):臨床試驗中使一方或多方不知道受試者治療分配的程序。單盲指受試者不知,雙盲指受試者、研究者、監查員或數據分析者均不知治療分配。 視察 (Inspection):有關的藥品監督管理部門對一項臨床試驗的文件、設備、記錄和其它方面進行官方審閱,視察可以在試驗單位、申辦者所在地或合同研究組織所在地進行。 弱勢對象(Vulnerable Subject):指受試者的意願受到不正當的影響,他們可能由於期望(無論正當與否)參加試驗而伴隨的利益,或者會因拒絕參加而受到上級的報復。有等級制度的成員,如醫學、藥學、齒科和護理專業的學生,附屬醫院和實驗室人員,製藥公司的僱員,軍人,以及被監禁的人。其他弱勢對象包括不治之症的患者,福利院的人,失業者或窮人,處於危急狀況的病人,少數民族,無家可歸者,流浪者,難民,未成年者,和那些無能力給出知情同意的人。 試驗方案(Protocol):敘述試驗的背景、理論基礎和目的,試驗設計、方法和組織,包括統計學考慮、試驗執行和完成的條件。方案必須由參加試驗的主要研究者、研究機構和申辦者簽章並註明日期。 試驗方案的修改(Protocol Amendment):對試驗方案的更改或正式澄清的書面描述。 試驗用藥 (Investigational Product):臨床試驗中用作試驗或對照藥品或安慰劑。 試驗單位(Trial site) : 真正開展與臨床試驗有關活動的場所。 試驗機構(Institution):實施臨床試驗的任何公共或私人的實體、代理機構、醫學或齒科設施。 監查報告(Monitoring Report):監查員到單位訪問後和/或其他與試驗有關的交流後,根據申辦者的標準操作規程寫給申辦者的書面報告。 監查員 (Monitor):由申辦者任命並對申辦者負責的具備相關知識的人員,其任務是監查和報告試驗的進行情況和核實數據,以保證臨床試驗按照方案、標準操作程序、《中成藥藥品臨床試驗質量管理規範》及其他相關法例的要求進行。 有關的藥品監督管理部門(Regulatory Authorities):有權進行管理的機構。試驗數據呈交予的一方,並會組織視察的藥品監督管理部門。 對照藥品(Comparator(Product)):臨床試驗中用作對照的試驗用藥品或市售藥品(即陽性)對照或安慰劑。 相關的法例要求(Applicable Regulatory Requirement) :有關實施試驗用29
40.
41.
42.
43.
44.
45.
46.
47.
48.
49.
50.
51. 藥品臨床試驗的任何法例。 稽查 (Audit):指由不直接涉及試驗的人員所進行的一種系統性檢查,以判斷試驗的實施、數據的記錄及分析是否與試驗方案、《中成藥藥品臨床試驗質量管理規範》與法規要求相符。 稽查軌跡(Audit Trail):允許重現事件過程的文件。 稽查報告(Audit Report) : 申辦者方的稽查員對稽查結果的書面評價。 稽查證書(Audit Certificate) : 稽查員確認已進行過稽查的聲明。 質量控制 (Quality Control):用以保證與臨床試驗相關活動的質量達到要求的操作性技術和規格。 標準操作規程 (Standard Operating Procedure, SOP):為有效地實施和完成某一臨床試驗中每項工作所擬定的標準和詳細的書面規程。 獨立的數據監查委員會(Independent Data-Monitoring Committee (IDMC)):申辦者方設立的一個獨立的數據監查委員會,定期對研究進展、安全性數據和重要的有效性參數進行評估,向申辦者建議是否繼續、修改或停止試驗。 隨機化(Randomization):為了減少偏倚,採用機遇決定將受試者分配到治療或對照組的過程。 臨床試驗(Clinical Trial):指任何在人體(病人或健康志願者)進行藥品的系統性研究,以證實或揭示試驗用藥品的作用、不良反應及/或試驗用藥品的吸收、分布、代謝和排泄,目的是確定試驗用藥品的療效與安全性。 臨床試驗中期報告(Interim Clinical Trial/Study Report):在試驗進行過程中得出的中期結果及分析後得出的評價報告。 藥品不良反應 (Adverse Drug Reaction):在按規定劑量正常應用藥品的過程中產生的有害而非所期望的、與藥品應用有因果關係的反應。在一種新藥或藥品新用途的臨床試驗中,其治療劑量尚未確定時,所有有害而非所期望的、與藥品應用有因果關係的反應,也應視為藥品不良反應。 嚴重不良事件 (Serious Adverse Event):臨床試驗過程中發生需住院治
療、延長住院時間、傷殘、影響工作能力、危及生命或死亡、導致先天畸形等事件。
30
附錄三
臨床試驗保存文件
一. 臨床試驗準備階段
臨床試驗保存文件 1 2 3 4 5 6 7
研究者手冊
試驗方案及其修正案(已簽名) 病例報告表(樣本)
研究者 保存 保存原件 保存
申辦者 保存 保存 保存 保存 保存 保存 保存
知情同意書及其他提供予受試者的書面資料保存原件 財務規定 保險陳述(必要時)
保存 保存
多方協議(已簽名)(研究者、申辦者、合同研保存 究組織)
8 倫理委員會批件(包括方案及其修正案、病例保存原件 報告表(如適用)、知情同意書及其他提供予受試者的書面資料、對受試者的補償(如有))
保存
9 10 11 12 13
倫理委員會成員表 臨床試驗申請表 臨床前實驗室資料
保存原件
保存 保存原件 保存原件 保存原件 保存原件
有關的藥品監督管理部門對試驗方案的批件保存 研究者履歷及相關文件
保存
31
14 15 16 17 18 19 20 21 22
臨床試驗有關的實驗室檢測正常值範圍 醫學或實驗室操作的質控證明 試驗用藥的標籤
試驗用藥與試驗相關物資的處理指南 試驗用藥與試驗相關物資的運貨單 試驗用藥的藥檢證明 設盲試驗的破盲規程 總隨機表
監查報告(試驗前及試驗開始時)
保存 保存原件 保存 保存
保存 保存 保存原件 保存 保存 保存原件 保存原件 保存原件 保存原件
二. 臨床試驗進行階段
臨床試驗保存文件 23 24
研究者手冊更新件
研究者 保存
申辦者 保存 保存
其他文件(保存 書面情況通知)的更新
25 倫理委員會對以上資料的更新的書面批准保存原件 (並註明日期)
保存
26 有關的藥品監督管理部門對試驗方案的修改保存(必要保存原件 的批件
時)
32
27 28 29 30 31 32 33 34 35 36 37 38 39
新研究者的履歷 保存 保存原件 保存 保存 保存 保存原件 保存原件 保存
保存原件 保存原件 保存 保存原件
醫學、實驗室檢查、操作的正常值範圍更新保存 醫學或實驗室操作的更新
試驗用藥品與試驗相關物資的運貨單 新批號試驗用藥的藥檢證明 監查員訪視報告
現場訪問之外的相關通訊聯絡紀錄 已簽名的知情同意書 原始醫療文件
病例報告表(已填寫,簽名,註明日期) 病例報告表的更正紀錄
研究者致申辦者的嚴重不良事件報告
保存原件 保存 保存 保存原件 保存原件 保存副本 保存副本 保存原件
申辦者致有關的藥品監督管理部門、倫理委保存 員會的未預期的嚴重藥品不良反應報告
40 41 42 43 44
申辦者向研究者通報的安全性資料 臨床試驗中期或年度報告 受試者的識別代碼表 受試者篩選表與入選表 試驗用藥登記表
保存 保存 保存原件 保存 保存
保存 保存 保存 保存
33
45 46
研究者簽名樣本
保存體液/組織樣本的紀錄
保存 保存
保存 保存
三. 臨床試驗完成後 臨床試驗保存文件 47 48 49 50 51 52 53
試驗用藥在試驗場地的管理責任 試驗用藥銷毀證明 完成試驗受試者編碼目錄 稽查證書(如需要) 最終監查報告 治療分配與破盲證明
研究者試驗完成報告(致倫理委員會、有關的藥品監督管理局) 54
研究者 保存 保存 保存
申辦者 保存 保存 保存 保存原件 保存原件 保存原件 保存原件
總結報告 保存 保存原件
34