医学遗传学
第一章 绪论
1、遗传病的特点:
第一、遗传病是垂直传播的,不同于传染病的水平传播。
第二、遗传病的患者在亲祖代与子孙中是以一定数量比例出现的,患者与正常者有一定的数量关系。
第三、遗传病是先天性的但不是所有的先天性疾病都是遗传病(如孕妇妊娠时风疹感染在成患儿的先天性心脏病)同时也不是所有的遗传病都在出生时都表现出来(如亨廷顿氏病)
第四、遗传病往往呈现出家族聚集性。但不是所有的有家族聚集性的疾病都是遗传病(如某些与饮食习惯有关的疾病)。 第五、遗传病的传染性。由于朊病毒的发现,现代遗传病的概念得到了进一步的拓展。PrP基因的突变会影响蛋白质的构象称为蛋白折叠病。错误折叠的蛋白可以诱导正常蛋白的变化所以也具有传染性。故从这个角度来讲遗传病也有传染性。
2、遗传病的分类可以分为:
1. 单基因病
? 常染色体显性 AD
? 常染色体隐性 AR
? 性染色体显性 XD
? 性染色体隐性 XR
2. 多基因病
3. 染色体病
4. 体细胞遗传病(这类疾病包括恶性肿瘤、自身免疫缺陷、衰老等。传统意义上的遗传病不包括这种)
5. 线粒体疾病
第三章 基因突变
1、一切生物细胞内的基因都能保持其相对稳定性,但在一定内外因素的影响下,遗传物质就可能发生变化,这种遗传物质的变化及其所引起的表型改变称为突变(mutation)。
2、基因突变的特征:多向性(同一基因座上的基因可独立发生多次不同的突变而形成复等位基因)、重复性、随机性、稀有性(在自然状态下发生突变的频率很低)、可逆性(可以发生回复突变)、有害性、突变(多数是有害的)
3、基因突变可以分为:自发突变、诱发突变。增加突变率的物质称为诱变剂。
4、诱变因素:
1. 物理因素
a) 紫外线(嘧啶二聚体,光复活修复(photoreactivation repair),哺乳动物没有)
b) 电离和电磁辐射(DNA链的断裂与染色体链的断裂;染色体重排、染色体结构改变)所引起的修复为: ? 超快修复:修复速度极快,在适宜条件下,大约2分钟内即可完成修复。
? 快修复:一般在X线照射后数分钟内,即可使超快修复所剩下的断裂单链的90%被修复。
? 慢修复:是由重组修复系统对快修复所不能修复的单链断裂加以修复的过程。一般修复时间较长。 c) 高温严寒(据王亚馥的《遗传学》所讲是对染色体倍性的影响)(可信性值得怀疑)
2. 化学因素
a) 羟胺(hydroxylamine,HA)碱基颠换
b) 亚硝酸或含亚硝基化合物 脱氨基从而导致碱基错配
c) 碱基类似物 代替碱基插入导致错配(5-溴尿嘧啶,EB)
d) 芳香族化合物 插入导致碱基移码突变(丫啶类,焦宁类)
e) 烷化剂 高度诱变活性(引起错配)
f) 烧烤兴奋剂也会有影响
3. 生物因素
1
a) 病毒 如风疹、麻疹、流感、疱疹等(分为DNA病毒和RNA病毒,前者的致病机理尚不知晓后者多半
由于逆转录的cDNA分子的插入)
b) 真菌和细菌 所产生的毒素(如黄曲霉素)
5、基因突变的形式与分子机制:
1. 静态突变(static mutation)是在一定条件下生物各世代中以相对稳定的频率发生的基因突变。可分为点突变
和片段突变。
a) 点突变(point mutation)DNA链中一个或一对碱基发生的改变
i. 碱基替换(base substitution)
1. 转换(transition) 嘌呤变嘌呤,嘧啶变嘧啶
2. 颠换(transvertion): 嘌呤变嘧啶,嘧啶变嘌呤
或
1. 同义突变(same sense mutation)
2. 无义突变(non-sense mutation)
3. 错义突变(missense mutation)镰刀性贫血(B链第六位aa,Glu变成了Val,GAG到GTG的
一个颠换)
4. 终止密码突变(terminator codon mutation)发生了通读
ii. 移码突变(frame-shift mutation)移码突变根据影响不同可以分为插入三个碱基序列(在第一个和第
三个插入的序列之间是不正常的,之前和之后的序列是正常的)和插入1或2个碱基序列。
b) 片断突变 DNA短小片断的重复、缺失、重排等
另外,突变还分为发生在编码区和发生在非编码区的突变(即影响非密码子区域的突变。影响非密码子区域的突变包括:调控序列突变(影响转录效率)和内含子与外显子剪辑位点突变(影响mRNA的加工,如GT-AG区域的改变)。
2、动态突变(dynamic mutation):串联重复的三核苷酸序列随着世代传递而拷贝数逐代累加的突变方式。三核苷酸重复扩增病TREDs。TREDs分为TRED1型(扩增发生在编码区)和TRED2型(扩增发生在非编码区)如: ? FRAX 脆性X综合症 X连锁
Xq27.3内(CGG)n重复数:60-200,正常:6-60
症状:智能低下,皮肤松弛,关节过度伸展,长脸。
? Huntington 舞蹈病 AD
? SBMA 脊髓肌萎缩 X连锁
雄激素受体蛋白,运动神经元受损
DNA的修复:(这里所讲的和生化书上的有一定的出入,请大家注意)
1. 光复活修复:应用于嘧啶二聚体;光复活酶参与;哺乳动物没有。
2. 切除修复(excision repair)貌似是NER的意思。但这里所讲的还是切除嘧啶二聚体。而且步骤和酶都不大
一样。(不是重点)
3. 重组修复(recombination repair)解决一个DNA链发生了结构改变(如嘧啶二聚体)而不能修复的问题。跳
过错误位点,子链完全正常。(也不是重点)
4. 电离辐射引起的DNA损伤的修复(见上文)
修复异常导致的遗传病:
? 着色性干皮病(XP)光修复及切除修复系统异常 解旋酶、核酸内切酶等修复蛋白的基因突变(其
实没有光修复的。。。。。。。)(XP家族蛋白的异常)
症状:对光敏感,皮肤、眼、舌易受损;皮肤上皮鳞状细胞或基底细胞皮肤癌;伴性发育不良、生长迟
缓、神经系统异常而学习能力差
? Bloom syndrome光敏感性reqQ解旋酶家族基因突变
2
症状:身材矮小、免疫功能低下、日光敏感性面部红斑和轻度颜面畸形
第四章基因突变的细胞分子生物学效应
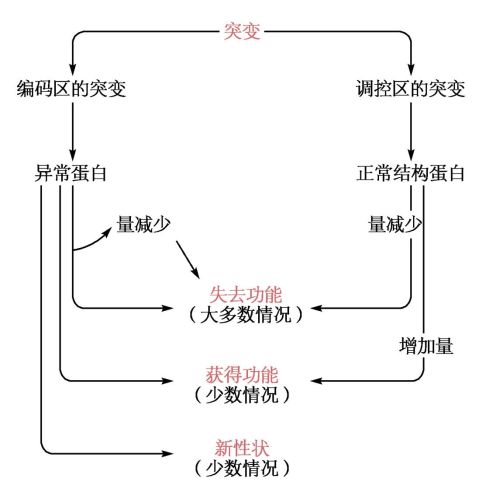
1、突变导致蛋白质功能异常:
a) 影响功能蛋白质的正常合成
i. 原发性损伤。如:β-珠蛋白生成障碍性贫血
机制:点突变导致转录受阻β-珠蛋白生成减少
ii. 继发性损伤。如:急性间隙性卟啉症(acute intermittent porphyria,AIP)机制:缺乏PBG脱氨酶使细
胞内ALA、胆色素原不能转化为血红素,血红素含量下降;而血红素的下降则调节着ALA合成酶表达的增加,ALA和胆色素原更严重的积聚,导致疾病。(临床表现:青春期以后出现神经系统症状)
b) 蛋白质正常结构的改变
i. 原发性损伤。如:Huntington 舞蹈病 PrP蛋白的堆积。
ii. 继发性损伤。如:Ehlers-Danlos综合征赖氨酸羟化酶缺陷所致,胶原分子上的赖氨酸不能被羟化,使胶
原分子间的连结发生障碍,而不能适应于组织细胞内胶原网络结构的形成,最终而导致结缔组织的结构改变和功能紊乱。
c) 蛋白的正常亚细胞定位
i. 原发性损伤。如:甲基丙二酸尿症 甲基丙二酰辅酶A羧基变位酶基因突变,使其不能进入线粒体。线
粒体内的甲基丙酰CoA因此不能转变为琥珀酰CoA,在线粒体内堆积而发病。
ii. 继发性损伤。如:I-细胞病(I-细胞即为包涵体细胞) 患者具有多种临床效应,包括骨骼异常、严重的
生长迟缓和智力低下等。M-6-P的形成出问题造成酸性水解酶的堆积。
d) 影响辅基或辅助因子与蛋白质结合、解离的突变
i. 原发性损伤。如:同型胱氨酸尿症。本病的分子缺陷是由于基因缺陷而致胱硫醚合成酶与辅助因子磷酸
吡哆醛的结合障碍而失去活性。大剂量的吡哆醛(维生素B6)具有一定的治疗作用
ii. 继发性损伤。
e) 影响蛋白质与其功能性亚基及其他因子之间结构组成关系
i. 原发性损伤。如:成骨不全症 症状:蛋白亚单位亲和力减低,如Ⅰ型胶原组装异常致使骨发育不良。 ii. 继发性损伤。如:Zellweger综合征(脑-肝-肾综合症)
2、突变导致蛋白产生的异常功能效应
a) 功能的丢失
b) 功能的加强。如:Von willebrand 病 (联系组胚中内皮细胞的W-P小体)vWF与血小板结合的功能加强,
不易从血小板上分离,患者损伤时,带有vWF的血小板的凝血作用减弱。
c) 新功能的出现
3
3、突变导致组织细胞蛋白表达类型的改变
a) 奢侈蛋白的突变。一般具有局限性,但有的可以影响全身。(如:苯丙酮尿症 PKU)
b) 组织特异性蛋白(tissue-specific protein)突变。一般情况下,组织特异性蛋白的突变所引起的病理生理改变
常局限于原发的特定的组织内部。
c) 持家蛋白的突变。持家蛋白突变所引起的临床效应通常局限在一个或几个持家蛋白起特殊作用的组织中。(前
面还有一句话是:一旦发生普遍性的突变后果不堪设想往往是致死性的。所以。。。。。)如:精氨琥珀酸裂解酶的异常(主要表现在鸟氨酸循环上而不是核酸合成上)
4、突变蛋白分子细胞病理学效应与相应临床表型之间的关系
a) 同一基因的不同突变产生不同的临床表型。同一单基因(基因座)的不同突变产生极其不同的临床表型意味
着遗传异质性(等位基因异质性)与临床异质性之间存在着因果联系。
b) 突变引发未能预测的临床效应。如:次黄嘌呤鸟嘌呤磷酸核糖基转移酶(HGPRT)遗传性缺陷导致自残综
合症
5、分子生物学机制
1) 先天性代谢病(遗传性酶病)
a) 基因突变引起酶分子的异常
i. 结构基因突变引起的酶结构异常 ①酶完全失去活性;②酶具一定程度的活性,但稳定性降低,容
易被迅速裂解而失去活性;③酶与底物的亲和力降低,无法有效结合;④酶蛋白与辅助因子的亲和
力下降,影响正常活性。
4
ii. 调节基因突变引起酶蛋白合成异常
b) 酶分子异常引起的代谢缺陷
i. 酶缺陷造成代谢底物缺乏 如:色氨酸加氧酶缺乏症
ii. 酶缺陷导致代谢底(产)物的堆积
1. 堆积产物对机体的直接危害 如:半乳糖血症
2. 堆积底(产)物激发代谢旁路开放。 如:PKU
iii. 酶缺陷导致代谢终产物的缺乏 如:白化病
iv. 酶缺陷导致反馈调节失常 如:21-羟化酶的缺陷导致先天性肾上腺皮质增生症(男婴出生时即为假
性性早熟,女婴生殖器异常后来逐渐男性化)
2) 分子病非酶蛋白分子结构和数量的异常所引起的疾病,统称为分子病(molecular disease)。如:运输蛋白、
免疫蛋白、膜载体蛋白、受体蛋白发生异常。
第五章、单基因疾病的遗传Monogenic Inheritance
1、定义:疾病的发生主要受一对等位基因控制,它们的传递方式遵循孟德尔遗传律。
①常染色体遗传
常染色体显性遗传
常染色体隐性遗传
②X伴性遗传
X连锁显性遗传
X连锁隐性遗传
③Y连锁遗传
2、系谱 (pedigree)
从先证者入手,追溯调查其所有家族成员(直系亲属和旁系亲属)的数目、亲属关系及某种遗传病(或性状)的分布等资料,并按一定格式将这些资料绘制而成的图解。
先证者(proband)
是某个家族中第一个被医生或遗传研究者发现的患某种遗传病的患者或具有某种性状的成员。
5
3、各类单基因遗传病的特点:
1) 常染色体完全显性遗传的特征
a) 由于致病基因位于常染色体上,因而致病基因的遗传与性别无关,即男女患病的机会均等
b) 患者的双亲中必有一个为患者,但绝大多数为杂合子,患者的同胞中约有1/2的可能性也为患者;
c) 系谱中可见本病的连续传递,即通常连续几代都可以看到患者;
d) 双亲无病时,子女一般不会患病(除非发生新的基因突变)。
例如:家族性高胆固醇血症、急性间歇性卟啉症、成骨不全、神经纤维瘤、多发性家族性结肠息肉、Α珠蛋白生成障碍性贫血、肌强直性营养不良、蜘蛛样指综合症、短指
2) 常染色体隐性遗传的特征
a) 由于基因位于常染色体上,所以它的发生与性别无关,男女发病机会相等;
b) 系谱中患者的分布往往是散发的,通常看不到连续传递现象,有时在整个系谱中甚至只有先证者一个患者; c) 患者的双亲表型往往正常,但都是致病基因的携带者,此时出生患儿的可能性约占1/4,患儿的正常同胞中
有2/3的可能性为携带者;
d) 近亲婚配时,子女中隐性遗传病的发病率要比非近亲婚配者高得多。这是由于他们来自共同的祖先,往往具
有某种共同的基因。
6
例如:镰状细胞贫血、婴儿黑朦性白痴(糖原储积症)(智力 肌无力)、β-地中海贫血、同型胱氨酸尿症、苯丙酮尿症、尿黑酸尿症、Friedreich家族性共济失调、半乳糖血症、肝豆状核变性(铜离子通道)、粘多糖累积症I型
3) X连锁显性遗传特点:
a) 人群中女性患者比男性患者约多一倍,前者病情常较轻;
b) 患者的双亲中必有一名是该病患者;
c) 男性患者的女儿全部都为患者,儿子全部正常
d) 女性患者(杂合子)的子女中各有50%的可能性是该病的患者;
e) 系谱中常可看到连续传递现象,这点与常染色体显性遗传一致。
例如:口面指综合征、色素失调、I型高氨血症、I型(鸟氨酸氨甲酰基转移酶缺乏)、Alport综合征色素失调症、抗维生素D佝偻病
4) X连锁隐性遗传病的遗传特点:
a) 人群中男性患者远较女性患者多,系谱中往往只有男性患者;
b) 双亲无病时,儿子可能发病,女儿则不会发病;儿子如果发病,母亲肯定是一个携带者,女儿也有1/2的可
能性为携带者;
c) 男性患者的兄弟、外祖父、舅父、姨表兄弟、外甥、外孙等也有可能是患者;
d) 如果女性是一患者,其父亲一定也是患者,母亲一定是携带者。
例如:色盲、鱼鳞癣、Lesch-Nyhan综合征、眼白化病、Hunter综合征、无丙种球蛋白血症、Fabry病(糖鞘脂贮积症)、Wiskott-Aldrich综合征、G-6-PD缺乏症、肾性尿崩症、慢性肉芽肿病、血友病A
5) Y连锁遗传病的遗传特点:具有Y连锁基因者均为男性,这些基因将随Y染色体进行传递,父转子、子传孙,
因此称为全男性遗传。外耳道多毛
4、影响单基因遗传病分析的因素
1) 不完全显性遗传 incomplete dominace 如:PTC味盲
2) 共显性(codominance)无显隐之分如:ABO血型
3) 延迟显性 delayed dominance杂合子在生命的早期,因致病基因并不表达或虽表达但尚不足以引起明显的临床表
现,只在达到一定的年龄后才表现出疾病,这一显性形式称为延迟显性。如:Huntington舞蹈病
4) 不规则显性遗传 irregular dominace杂合子的显性基因由于某种原因而不表现出相应的性状,因此在系谱中可以
出现隔代遗传的现象。不表现出显性性状的个体称顿挫型。外显率是一定环境条件下,群体中某一基因型(在杂合状态下)个体表现出相应表型的百分比。分为不完全外显和完全外显如:多指
5) 表现度 expressivity表现度是基因在个体中的表现程度,或者说具有相同基因型的不同个体或同一个体的不同部
位,由于各自遗传背景和环境因素的不同,所表现的程度可有显著的并异。外显率与表现度的区别在于一个质与量的区别。外显率是在讨论是否有,表现度是在讨论已经有了那么表现到了一个什么程度。
6) 基因多效性 genetic pleiotropy一个基因可以决定或影响多个性状。如:半乳糖血症表现出智能低下/黄疸、腹水、
肝硬化/白内障;GPTU肝豆状核变性表现出肝损伤/神经异常。
7) 遗传异质性 genetic heterogeneity一种遗传性状可以由多个不同的基因控制/遗传改变引起。包括:基因座异质
性和等位基因异质性。例如对于智能低下: Tay-Sachs/PKU/半乳糖血症、肌营养不良(最为极端的是闭眼肌麻痹不能闭眼,患者最后死于呼吸肌麻痹等)
8) 同一基因可产生不同突变:同一基因的不同突变可引起显性或隐性遗传病。如:β珠蛋白基因突变导致地中海贫
血其中分为:127谷氨酰胺→脯氨酸,β+-Houston-地中海贫血AD和26谷氨酸→赖氨酸,β+-E-地中海贫血, AR
9) 遗传早现 anticipation遗传早现是指一些遗传病(通常为显性遗传病)在连续几代的遗传中,发病年龄提前而
且病情严重程度增加。如:脊髓小脑性共济失调1型 6p CAG
10) 遗传印迹 genetic imprinting 越来越多的研究显示一个个体的同源染色体(或相应的一对等位基因)因分别来自
其父方或母方,而表现出功能上的差异,因此当当它们其一发生改变时,所形成的表型也有不同,这种现象称为遗传印记或基因组印记(genomic imprinting)、亲代印记(parental imprinting)。 例如:Huntington舞蹈病 母亲传递不呈早发现象而父亲传递则呈早发现象
7
11) 从性遗传 sex-conditioned inheritance 位于常染色体上的基因,由于性别的差异而显示出男女性分布比例上的差
异或基因表达程度上的差异。如:秃顶
12) 限性遗传 sex-limited inheritance 常染色体上的基因,由于基因表达的性别限制,只在一种性别表现,而在另一
种性别则完全不能表现。如:女性的子宫阴道积水症,男性的前列腺癌等。
13) X染色体失活 X-chromosome inactivation 即Lyon假说,女性两条X染色体在胚胎发育早期就随机失活了其中
的一条,因此女性的两条X染色体存在嵌合现象。
14) 拟表型 phenocopy由于环境因素的作用使个体的表型恰好与某一特定基因所产生的表型相同或相似,这种由
环境因素引起的表型称为拟表型。例如:先天性聋哑/药物致聋哑
第六章 多基因疾病的遗传 Polygenic Inheritance
1、定义:一些性状(数量性状)与疾病不是由一对基因所控制的而是由多对基因所控制的,性状的遗传不遵守孟德尔遗传规律,受环境影响大。称为:数量性状或是多基因性状。遗传方式为多基因遗传(polygenic inheritance)或是多因子遗传(multifactorial inheritance, MF)
2、单基因遗传 群体患病率低 万分之一
多基因遗传 群体患病率高 0.1%-1%
3、微效基因(minor gene)人类的一些遗传性状或某些遗传病的遗传基础不是一对主基因,而是几对基因,每一对基因对遗传性状或遗传病形成的作用是微小的。
累加效应 additive effect 累加基因共显
多基因遗传病 性状或疾病的遗传方式取决于两个以上微效基因的累加作用,还受环境因子的影响,因此这类性状也称为复杂性状或复杂疾病(complex disease)。
主基因 各微效基因所发挥的作用可能并不相同,存在起主要作用的主基因。
质量性状(qualitative character) 基因型表型关系直接,变异群体分布不连续。
数量性状(quantitative character)性状连续变异,相邻个体间差异很小。呈正态分布。单峰原因:多对微效基因;基因随机组合。
易患性(liability)在多基因遗传病发生中,遗传因素和环境因素(与易感性的区别)共同作用决定一个个体患某种遗传病的可能性称为易患性。
易感性(susceptibility)易感性特指由遗传因素决定的患病风险,仅代表个体所含有的遗传因素;但在一定的环境条件下,易感性高低可代表易患性高低。
发病阈值 (threshold)当一个个体易患性(内含环境因素)高到一定限度就可能发病。这种由易患性所导致的多基因遗传病发病最低限度称为发病阈值。 与线粒体中遗传发病阈值的区别。
遗传度(heritability)多基因累加效应对疾病易患性变异的贡献大小。遗传度愈大,表明遗传因素对病因的贡献愈大。
4、常见多基因遗传病:近视、高血压、糖尿病、精神分裂症、哮喘
5、影响多基因遗传病再发风险估计的因素
1) 患病率与亲属级别有关
2) 患者亲属再发风险与亲属中受累人数有关
3) 患者亲属再发风险与患者畸形或疾病严重程度有关
4) 多基因遗传病的群体患病率存在性别差异时,亲属再发风险与性别有关。比如:群体患病率较低即阈值较高
的那种性别罹患,则患者亲属的发病风险较高。
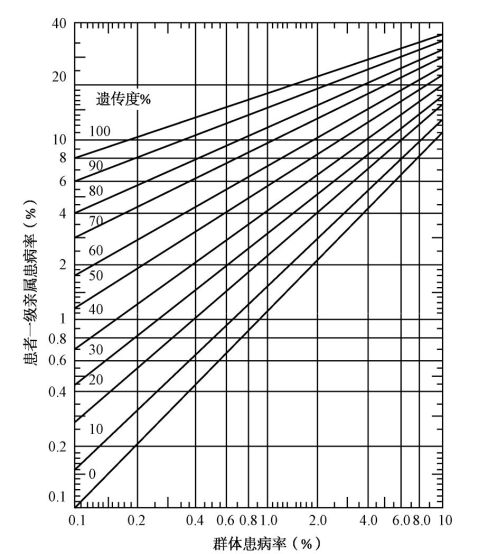
6、计算一级亲属患病率的方法:
8
无脑畸形和脊柱裂的患病率为0.38%,在图中横轴上查出0.38之点,作一垂直线与纵轴平,已知此病的遗传度为60%,从图中找出遗传度60%的斜线,把它和0.38的垂直线相交点作一横线在纵轴上的一点近于4,即表明该病的一级亲属患病率接近4%。
第八章 线粒体疾病的遗传 Inheritance of Mitochondrail diseases
1、线粒体基因组 线粒体内含有DNA分子,被称为人类第25号染色体,是细胞核以外含有遗传信息和表达系统的细胞器,其遗传特点表现为非孟德尔遗传方式,又称核外遗传。
2、线粒体基因组特点:
1) 线粒体基因组全长16569bp;
2) 不与组蛋白结合,呈裸露闭环双链状,根据其转录产物在CsCl中密度的不同分为重链和轻链;
3) 重链(H链)富含鸟嘌呤,轻链(L链)富含胞嘧啶。
3、线粒体基因组构成
1) mtDNA分为编码区与非编码区:编码区各基因之间排列极为紧凑,除与 mt DNA 复制及转录有关的一小段
区域外,无内含子序列。部分区域出现重叠,缺少终止密码子,仅以U或UA结尾。一共37个基因2个mtrRNA,22个mttRNA,13个与氧化磷酸化相关的蛋白。(OXPHOS)
2) 非编码区:(D loop),1122bp,H链复制起始点,H链和L链转录启动子,和4个保守序列; L链复制起始
区。
4、半自主性体现在:
1) mtDNA仅编码13种,其他绝大部分蛋白质亚基和其他维持线粒体结构和功能的蛋白质都依赖于核
DNA
9
(nuclear DNA,nDNA)编码,在细胞质中合成后,经特定转运方式进入线粒体;
2) mtDNA基因的表达受nDNA的制约,线粒体氧化磷酸化系统的组装和维护需要nDNA和mtDNA的协调,
二者共同作用参与机体代谢调节。
5、线粒体DNA的复制是D环复制也是半保留复制。
6、转录特点: 两条链均有编码功能
1) 两条链从D-环区的启动子处同时开始以相同速率转录,L链按顺时针方向转录,H链按逆时针方向转录
2) mtDNA的基因之间无终止子
3) tRNA基因通常位于mRNA基因和rRNA基因之间
4) mtDNA的遗传密码与nDNA不完全相同
5) 线粒体中的tRNA兼用性较强
7、线粒体疾病的机制:线粒体疾病是由于各种原因使 mt DNA 或 / 和nDNA 发生基因突变,线粒体内酶功能缺陷,ATP 合成障碍,不能维持细胞的正常生理功能产生氧化应激,使氧自由基产生增加,诱导细胞凋亡。
8、突变率高的原因:
1) mtDNA中基因排列紧凑,任何突变都可能会影响到其基因组内的某一重要功能区域
2) mtDNA是裸露的分子,不与组蛋白结合
3) mtDNA位于线粒体内膜附近,直接暴露于呼吸链代谢产生的超氧离子和电子传递产生的羟自由基中,极易
受氧化损伤
4) mtDNA复制频率较高,复制时不对称
5) 缺乏有效的DNA损伤修复能力
9、线粒体基因的突变类型
1) 点突变
2) 大片段重组:大片段的缺失往往涉及多个基因,可导致线粒体OXPHOS功能下降,产生的ATP减少,从而
影响组织器官的功能
3) mtDNA数量减少:AD或AR遗传,与nDNA有关
10、mtDNA的修复:切除修复与转移修复
11、mt遗传特点:(重点)
1) 母系遗传:双亲信息的不等量表现决定了线粒体遗传病的传递方式不符合孟德尔遗传,而是表现为母系遗传
(maternal inheritance),即母亲将mtDNA传递给她的儿子和女儿,但只有女儿能将其mtDNA传递给下一代。
2) 异质性:如果同一组织或细胞中的mtDNA分子都是一致的,称为同质性(homoplasmy)。一些个体同时存
在两种或两种以上类型的mtDNA,这是由于mtDNA发生突变,导致一个细胞内同时存在野生型mtDNA和突变型mtDNA,称为异质性(heteroplasmy)。野生型mtDNA对突变型mtDNA有保护和补偿作用,因此,mtDNA突变时并不立即产生严重后果。分为序列异质性和长度异质性
3) 阈值效应 :mtDNA突变可以影响线粒体OXPHOS的功能,引起ATP合成障碍,导致疾病发生,但实际上
基因型和表现型的关系并非如此简单。 线粒体的异质性水平 + 最低能量引起特定组织器官功能障碍的突变线粒体的最少数量称为阈值。
4) 不均等的有丝分裂分离:细胞分裂时,突变型和野生型mtDNA发生分离,随机地分配到子细胞中,使子细
胞拥有不同比例的突变型mtDNA分子,称复制分离。
12、mt疾病:线粒体病是一组多系统疾病,因中枢神经系统和骨骼肌对能量的依赖性最强,故临床症状以中枢神经系统和骨骼肌病变为特征,对应线粒体脑病/线粒体肌病以及线粒体脑肌病。例如:Kearns-Sayre综合征(眼肌病)、Pearson-骨髓/胰腺综合征、线粒体心肌病、帕金森病、Alzheimer病、非胰岛素依赖型糖尿病
13、Leber遗传性视神经病(Leber hereditary optic neuropathy,LHON)是第一个被发现的母系遗传病
临床症状
主要症状为视神经退行性变,故又称Leber视神经萎缩。患者多在18~20岁发病,男性较多见,个体细
10
胞中突变mtDNA超过96%时发病,少于80%时男性病人症状不明显。临床表现为双侧视神经严重萎缩引起的急性或亚急性双侧中央视力丧失,可伴有神经、心血管、骨骼肌等系统异常,如头痛、癫痫及心律失常等。
遗传学
诱发LHON的mtDNA突变均为点突变。
如:G11778A、G14459A、G3460A、T14484C、G15257A
14、Kearns-Sayre综合征KSS、慢性进行性眼外肌瘫痪 CPEO 属于线粒体脑肌病
临床症状
患者可表现一系列不同的症状,从仅有眼肌麻痹、眼睑下垂及四肢肌病到视网膜色素变性、乳酸中毒、感觉神经性听力丧失、运动失调、心脏传导功能障碍,甚至痴呆。具前一症状时,称为CPEO,发展成为后一症状时,即称为KSS。
KSS和CPEO主要是由于mtDNA的缺失引起的,缺失类型多样,一般缺失长度为0.5~8kb,最常见的类型是5.0kb缺失;
缺失部位多发生在重链和轻链两个复制起始点之间,可抑制线粒体翻译,酶活性下降。
由于涉及多个基因的缺失,患者可出现不同程度的线粒体蛋白质合成缺陷,影响四种呼吸链复合体;
偶见8334位点(tRNALeu)和3242位点(tRNALeu)点突变可引起CPEO。
第九章 人类染色体human chromosome
1、染色体(chromosome) 是遗传物质(基因)的载体。它由DNA和蛋白质等构成,具有储存和传递遗传信息的作用。
2、染色质和染色体实质上是同一物质在不同细胞周期、执行不同生理功能时不同的存在形式。
3、Lyon假说 :
1) 失活发生在胚胎发育早期(人类晚期囊胚期);
2) X染色体的失活是随机的;
3) 失活是完全的;
4) 失活是永久的和克隆式繁殖的。
5) 局部失活:失活中心在Xq13,其大部分基因是失活的,但并非全部失活 。
6) 非随机失活:X染色体结构异常、X与常染色体平衡易位
4、染色体组(chromosome set) 一个正常生殖细胞(配子)中所含的全套染色体称为一个染色体组 基因组(genome) 一个染色体组所含的全部基因
5、人类染色体可分为:中着丝粒染色体(metacentric chromosome) 亚中着丝粒染色体(submetacentric chromosome) 近端着丝粒染色体(acrocentric chromosome)可以分为ABCDEFG七个组群
6、染色体显带:经不同的方法处理染色体,经染色后使染色体在纵轴上显示明、暗或着色深、浅相间的横纹即显带(Banding)。
7、核型分析里的常用符号
8、染色体畸变(chromosome aberration)是指体细胞或生殖细胞内染色体发生的异常改变。包括:数目畸变和结构畸变(deficiencies, duplications, translocations, inversions, etc)。
9、致畸因素:
1) 抗肿瘤药物、保胎及预防妊娠反应的药物、抗痉挛药物、
2) 工业毒物,如:苯、甲苯、铝、砷、二硫化碳、氯丁二稀、氯乙烯单体等,都可以导致染色体畸变。(甲醛、
镉)
3) 食品添加剂
4) 农药
5) 电离辐射、电磁辐、
6) 生物类毒素如:杂色曲霉素、黄曲霉素、棒曲霉素等;病毒如:风疹病毒、乙肝病毒、麻疹病毒和巨细胞病
毒等
11
7) 母亲年龄
10、整倍体改变的机制
1) 双雄受精
2) 双雌受精
3) 核内复制:在一次细胞分裂时,DNA不是复制一次,而是复制了两次,而细胞只分裂了一次。这样形成的
两个子细胞都是四倍体。
4) 核内有丝分裂:在正常的细胞分裂时,染色体正常复制了一次。但至分裂中期时,核膜仍未破裂、消失,也
无纺锤体的形成,因此,细胞分裂未能进入后期和末期,没有细胞质的分裂,结果细胞内含有四个染色体组,形成四倍体。
11、非整倍体的改变:
亚二倍体(hypodiploid) 体细胞中染色体数目少了一条或数条
超二倍体(hyperdiploid) 体细胞中染色体数目多了一条或数条
嵌合体(mosaic):一个个体内同时存在两种或两种以上核型的细胞系。(肿瘤细胞)
12、非整倍体改变的机制
1) 染色体不分离(non-disjunction)
a) 受精卵早期卵裂的有丝分裂不分离 (超二倍体、嵌合体)
b) 减数分裂时发生染色体不分离( 超二倍体、二倍体、亚二倍体)
2) 染色体丢失(chromosome lose)(亚二倍体、嵌合体)
第十一章 单基因病Single Gene Disorder
1、分子病(molecular disease) 分子病是指基因突变使蛋白质的分子结构或合成的量异常直接引起机体功能障碍的一类疾病。
1) 血红蛋白病(Hemoglobin , Hb Disease)
a) 血红蛋白病 ——珠蛋白结构异常 例如:点突变
i. 错义突变:(镰状细胞贫血,β链N端 6Glu→Val)
ii. 无义突变:(Hb Mckees Rorks变异型,β链 145UAU→UAA,C端少了2个氨基酸)
iii. 终止密码子突变:(Hb seal Rock变异型,α链UAA→GAA,多31个氨基酸)
iv. 移码突变:碱基缺失或插入(Hb Tak β链第147位终止密码子UAA前插入2个碱基AC)
v. 密码子缺失和插入: 组成某个密码子的碱基同时缺失或插入一个或多个密码子→肽链缺少或增加
了部分氨基酸→结构和功能异常
vi. 融合突变:Hb Lepore 变异型,基因δ和β发生错误联合和不等交换→δ链N端和β链的C端部
分融合→δβ链
主要的遗传效益:1、血红蛋白稳定性改变,多肽链构象改变,血红素所在位置的构象改变。
2、血红蛋白带氧能力降低。
例如:镰状细胞贫血症(sickle cell anemia, HbS) AR
临床表现
镰变细胞引起血粘性增加,易使微细血管栓塞,造成散发性的组织局部缺氧,甚至坏死,产生肌肉骨骼痛、腹痛等痛性危象。同时镰状细胞的变形能力降低,通过狭窄的毛细血管时,不易变形通过,挤压时易破裂,导致溶血性贫血
b) 珠蛋白生成障碍性贫血(地中海贫血)—— 珠蛋白合成速率改变
i. α珠蛋白生成障碍性贫血(αthalassemia)
遗传学 常染色体不完全显性
一条16号染色体上缺失一个α基因——α+地贫;缺失两个α基因——α0地贫
Hb Bart’s胎儿水肿综合症(α0/α0)
12
? 血红蛋白H病(α0/α+)
? 标准型(轻型)α地中海贫血(α0/αA 或 α+/α+ )
? 静止型α地中海贫血(α+/αA )
ii. β珠蛋白生成障碍性贫血(β-thalassemia)
遗传学 AR
完全不能合成β 链—— β0地贫;能部分合成β 链——β+ 地贫(约为正常的5% ~ 30%)
? 重型β 地中海贫血(β0 / β0 及 β0 / β+ )患者不能合成β链,α链过剩而沉降
到红细胞膜上,引起膜的性能改变,发生严重的溶血反应。组织缺氧,促进红细胞生成素分泌,
刺激骨髓增生,骨质受损变得疏松,可出现鼻塌眼肿、上颔前突、头大额隆等特殊的“地中海
贫血面容”
? 中间型β 地中海贫血(β+/ β+及 β+ / δβ+ ):一般是β+地中海贫血基因的纯合
子,患者的基因型通常为β+(高F)/β+(高F)或β+/δβ+。病人的症状介于重型和轻
型之间,故称为中间型。
? 轻型β 地中海贫血(β0 / βA及 β+ / βA ):发生于β0或β+地中海贫血基因的杂
合子,无任何临床症状。
2) 血浆蛋白病(Plasma Protein Disease) 血浆蛋白病是血浆蛋白遗传性缺陷所引起的一组疾病。在血浆蛋白病中
以血友病较常见。血友病(hemophilia)是一类遗传性凝血功能障碍的出血性疾病
a) 血友病A(hemophilia , 甲型血友病或第Ⅷ因子缺乏症)
遗传学与发病机制
? XR
? FⅧ缺乏所致凝血缺陷
? FⅧAHG(抗血友病球蛋白,FⅧ凝血成分)
? FⅧAg(Ⅷ因子相关抗原)
? ⅧvWF(von Willebrand因子)
临床表现:
? 重型:出生后即发病,“自发性”肌肉、关节出血
? 中间型:发病年龄较早,出血倾向较明显
? 轻型:发病年龄较晚,无自主性出血,关节、肌肉出血较少
基因诊断:DNA印记杂交技术、PCR-RFLP技术、变性梯度凝胶电泳(DGGE)法、RNA酶裂解法、PCR-SSCP(单链构象多态性)法
b) 血友病B( Hemophilia B )
发病机制
凝血因子Ⅸ缺乏或其凝血功能降低所致(FⅨ基因定位于Xq27,全长35kb,由8个外显子和7个内含子组成,成熟的FⅨ由415个氨基酸构成。)
临床表现
大片段基因缺失→ 血浆中FⅨ抗原水平甚低或完全没有→ 严重的血友病B
点突变:无义突变→ 重型血友病B 错义突变→ 不同程度的血友病
基因诊断:RFLPs连锁分析法、Southern印迹杂交分析 ------大片段缺失
c) 血友病C( Hemophilia C)
d) Von Willebrand Disease(血管性假血友病, AD或AR)
vWF因子缺乏所致,
3) 结构蛋白缺陷病
a) 胶原旦白病(inherited disorders of collagen )
例如:成骨不全 (Osteogenesis Imperfecta)
13
遗传学
? 常染色体显性遗传
? Ⅰ型胶原异常所致
? 具有遗传异质性
Ehlers-Danlos综合症
遗传学
? 遗传异质性
? AD或AR
临床:指关节可以过度弯曲伸展。皮肤松弛、皮肤脆薄易于损伤伤后愈合差,疤痕菲薄
b) 肌营养不良症(muscular dystrophy)
例如:Duchenne 型肌营养不良症 (DMD,XR)
遗传学
DMD基因定位于Xp21.2,全长约2300kb,含70个外显子,编码400kd的多肽链-dystrophin;
5?端或中央区缺失突变 →dystrophin无法合成(DMD)或合成量降低(BMD)。
Becker型肌营养不良症 (BMD)
4) 受体病 (Receptor Disease)
例如:家族性高胆固醇血症 (familiar hypercholesterolemia)
遗传学
本病为常染色体显性遗传,LDL受体基因定位于19p13.1-p13.2。LDL基因突变包括碱基替换、插入、缺失
等,其中以碱基缺失较多见。
发病机制
? 无受体合成
? 合成后转运到细胞表面的过程缺陷
? 受体与LDL的结合缺陷
? 内运过程缺陷
? 在被凹聚集过程缺陷
临床:脂肪沉积于关节、眼睑等处。
5) 膜转运蛋白病
例如:囊性纤维样变 (cystic fibrosis , CF )(Cl-通道蛋白,CFTR)
发病机制和临床表现:
全身外分泌腺细胞分泌黏液不能及时清除 → →积滞在导管和腺泡中→ →阻塞和感染
累及呼吸道、消化道、汗腺、男性患者伴先天性双侧输精管缺如
胱氨酸尿症 ( cystinuria)
遗传学
Ⅰ型为常染色体隐性遗传;Ⅱ型和Ⅲ型均为常染色体不完全隐性遗传。
发病机制
膜转运蛋白缺陷 → →肾小管对氨基酸重吸收障碍 → →尿液中氨基酸含量升高→ →尿路结石 临床表现
尿路结石引起尿路感染和绞痛等症状
先天性葡萄糖、半乳糖吸收不良症(congenital glucose-galactose malasorption)
遗传学 AR
发病机制
小肠上皮细胞转运葡萄糖、半乳糖的膜载体蛋白异常,致使葡萄糖和半乳糖吸收障碍,患者肠道内渗透压改变而使肠液增加,患者出现水样腹泻。
14
临床表现
婴儿喂食含葡萄糖和半乳糖的食物后随着腹泻加重继而出现脱水、营养不良等症状。
2、先天性代谢缺陷(inborn errors of metabolism)
基因突变所引起的酶的结构改变或合成障碍,都有可能引起某种代谢过程的中断或紊乱。如果这种基因突变恰好发生在生殖细胞或受精卵中,就有可能传递给后代,从而使后代产生相应的先天性代谢缺陷(或遗传性酶病(hereditary enzymopathy)
3、先天性代谢缺陷的共同规律:
1) 酶缺陷与酶活性
2) 底物堆积和产物缺乏
3) 底物分子的大小与性质
4) 临床表型与酶缺陷
4、各论:
1) 糖代谢障碍
a) 半乳糖血症(galactosemia)
遗传学
半乳糖-1-磷酸尿苷酰转移酶(GPUT)缺陷,本病为常染色体隐性遗传,致病基因定位于9p13,发病率约为1/50000 。
发病机制
半乳糖和1-磷酸半乳糖在血中累积,部分随尿排出。1-磷酸半乳糖在脑、肾中累积可分别导致智力障碍、肝损伤甚至肝硬化及肾功能损伤等。
临床表现
主要表现为患儿对乳糖不耐受,婴儿哺乳后呕吐、腹泻,继而出现白内障、肝硬化、黄疸、腹水、智力发育不全等。
诊断
实验室检查
测定酶活性
血和尿中半乳糖浓度
红细胞中半乳糖-1-磷酸的含量
预防及治疗
确诊后应立即停乳,代之豆浆。严重者应逐日监测尿内半乳糖水平。
新生儿筛查可早期诊断。可用羊水细胞或绒毛细胞培养物的酶学分析行产前诊断。
b) 蚕豆病(葡萄糖-6-磷酸脱氢酶缺乏症)
遗传学
X连锁不完全显性,基因定位于Xq28,G6PD缺陷。分布具有世界性,我国主要分布在长江以南,尤其是广东。
发病机制
G6PD缺陷,导致GSH生成减少,细胞抗氧化损伤能力下降,引起红细胞膜损伤;血红蛋白β链93位半胱氨酸巯基氧化,四聚体解离,形成Heinz小体,红细胞变形能力下降,破坏出现溶血。
蚕豆、伯氨喹啉等富含氧化物,服用后可加重该病。
临床表现:
常见于10岁以下小儿,1—5岁为发病高峰。进食蚕豆后,蚕豆中的物质可使患儿体内的葡萄糖六磷酸脱氢酶被分解,导致大量红细胞破裂溶血,使患儿出现溶血性贫血。发病轻重则与吃蚕豆多少无关,一般在进食蚕豆后1—2天内发病,早期病人常出现全身不适、胃口不佳、发热、头昏等酷似肝炎的症状。重症病人会由于大量红细胞被破坏而释放出胆红素,导致黄疸、浓茶样的血红蛋白尿、肝脾肿大,并有恶心呕吐、腹痛等临床症状,
15
若不及时抢救治疗,发病后1—2天内就会死亡。
c) 糖原贮积症(glycogen storage disease,GSD)
遗传学
糖原贮积症是一类较罕见的遗传代谢病。由于酶的缺陷,使糖原在肝脏及肌肉中的代谢缺陷所致。根据所缺的酶不同,可将糖原贮积症分为Ⅰ~Ⅷ型,多数为常染色体隐性遗传,以Ⅰ型为最常见。
i. Ⅰ型糖原贮积症:
发病机制
葡萄糖-6-磷酸酶的基因缺陷,使肝、肾、及肠粘膜等组织中糖原蓄积。
临床表现
患者易出现低血糖,并有肝、肾肿大等症状,严重时会发生酸中毒。
ii. Ⅱ型糖原贮积症:
发病机制
基因定位于17q25.2,溶酶体内α-葡萄糖苷酶的缺乏,使糖原处理障碍,造成溶酶体内糖原堆积, 病变累及全身肌肉。
临床表现
一般在儿童期即发病,患者因心肌无力、心脏扩大而最终死于心力衰竭
d) 粘多糖贮积症(mucopolysaccharidosis,MPS)
发病机制
粘多糖由结缔组织合成,是二糖重复单位串联而成的多糖链,粘多糖分解时需要多种酶的参与,这些酶的遗传性缺陷可导致粘多糖降解受阻,蓄积于溶酶体中形成粘多糖贮积症。
临床:
患儿出现肝脾肿大、骨骼异常、智力障碍等症状,蓄积的粘多糖可随患儿的尿液排除
2) 氨基酸代谢障碍
a) 苯丙酮尿症(Phenylketonuria,PKU)
遗传学
一常染色体隐性遗传性氨基酸代谢病,疾病基因已定位于12q24.1。
发病机制
PKU患者PAH基因突变使患者肝脏内PAH缺乏,苯丙氨酸不能转变为酪氨酸,后者转化为苯丙酮酸和苯乳酸并在体内累积,并导致血液和尿液中苯丙氨酸及其衍生物排出增多。
临床表现
临床上表现为精神发育迟缓,皮肤、毛发和虹膜色素减退,头发呈赤褐色,癫痫,湿疹,特殊的鼠样臭味尿。患儿在出生后若不及早得到低苯丙氨酸饮食治疗,便出现不可逆的大脑损害和严重的智力发育障碍。
诊断:临床表现、新生儿筛查、尿三氯化铁试验、血氨基酸分析、酶学分析、DNA分析
治疗:目前临床上常在婴儿出生后立即进行PKU的筛查,一经肯定,立即给患儿停乳,喂给低苯丙氨酸水解蛋白,禁荤食、乳类、豆类和豆制品,可以达到临床痊愈。
b) 白化病(albinism)
遗传学
酪氨酸酶基因缺陷,遗传方式为AR;致病基因定位于11q14-q21。
发病机制
患者体内酪氨酸酶酶缺乏,不能有效地催化酪氨酸转变为黑色素前体,最终导致代谢终产物黑色素缺乏。
临床表现:全身白化;视网膜、无色素,畏光。
c) 尿黑酸尿症(alkaptonuria)
遗传学:本病呈常染色体隐性遗传, 疾病基因定位于 3q21-q23,患者尿黑酸氧化酶缺陷。
16
临床表现
病人的尿中含有尿黑酸(alkapton),曝光后可变为黑色的物质,这种病症在婴儿期就可表现出来,到成年时由于尿黑酸大量沉积于关节与软骨外,使关节变性。一般无明显的临床表现,严重时可出现关节炎,并发心脏病。
3) 核酸代谢障碍
a) 次黄嘌呤鸟嘌呤磷酸核糖转移酶缺陷症(Lesch-Nyhan sydrome)
遗传学 本病是一种由于次黄嘌呤鸟嘌呤磷酸核糖转移酶(HGPRT)缺陷所致的疾病,XR,基因定位于Xp26-p27.2。
发病机制
HGPRT是体内核酸补救合成途径的关键酶,它的缺陷使黄嘌呤、鸟嘌呤向相应核苷酸的转化
受阻,底物在体内堆积,特别是在神经系统中的堆积,进而引起发病。
临床表现
患者一般为男性,出生3-4个月开始出现神经系统症状,激惹不安,烦躁,运动发育迟缓;约
1岁后出现舞蹈样手足徐动,肌张力高,下肢呈剪刀样交叉;约2-3岁起,表现强迫性自我摧残行为,多数智商低于65;患儿精神发育迟滞,强直性大脑性瘫痪。
诊断:红细胞、皮肤成纤维细胞测定HGPRT酶活性、测晨尿的尿酸/肌酐比值可作筛选手段、利用羊水细
胞检查可行产前诊断
治疗:现尚无特殊治疗方法,一般采用口服黄嘌呤氧化酶的抑制剂—别嘌呤醇,可抑制尿酸的生成,防止
尿酸结石和肾脏病损,但不改善神经系统症状。饮食尽量无嘌呤摄入。
b) 着色性干皮病(xeroderma pigmentosum XP)
遗传学
着色性干皮病为常染色体隐性遗传病。由于患者体内缺乏核酸内切酶引起的疾病。本病可分为(XPA-XPG)7型,目前已克隆出XPA、XPB、XPC、XPD的基因,其中XPA定位于9q34.1, XPB定位于2q21。 发病机制
核酸内切酶缺乏→不能切除由紫外线诱发的嘧啶二聚体(特别是胸腺嘧啶二聚体,T-T)。
临床表现
患者皮肤对阳光过敏,日照后可出现红斑、水肿、色素沉着、干燥、角化过度及萎缩等皮损。有些病人智能落后,感音神经性聋及共济失调。易患基底细胞癌、鳞癌、恶性黑色素瘤等,均伴有免疫系统的异常。
4) 抗胰蛋白酶缺乏症 (α1-antitrypsin )
遗传学
α1抗胰蛋白酶为糖蛋白,基因定位于14q32.1 ,本病遗传方式为AR。
发病机制
α1抗胰蛋白酶存在于血浆中,尿液、支气管分泌物等,可抑制血清中多种蛋白酶活性。
α1-抗胰蛋白酶缺乏症特征是血清中α1-AT水平下降。
第十二章多基因遗传疾病 Polygenetic disorders
1、定义:一些常见病或多发畸形,有家族聚集现象,但患者同胞发病远低于1/2或1/4,发病还受到环境因素的影响。这种发病有一定多基因遗传基础的一类复杂疾病,称为多基因遗传病。
2、相关疾病
1) 精神分裂症(schizophrenia , SP)
临床特征
? 联想障碍
? 情感淡漠、情感不协调
? 意志活动减退、缺乏
17
? 幻觉、妄想和紧张
? 缺乏自知力
遗传:
SP与遗传有关的依据:
? 患者子女患病机率为35%一68%,正常人群仅为0.86%一1%。
? 遗传度:70%-80%
? 同卵双生发病一致率比异卵双生发病一致率高4-6倍
易感基因
? DRD基因
? 5-HTRA2
? HLA(6p21.31)
? 某些SP患者存在自身免疫现象,推测HLA可能参与精神分裂症的发病过程;
? 研究证实HLA-A1、A2、A9、B5、CW4、DR8精神分裂症呈正相关;
? HLA-DR4、DQB1 与精神分裂症呈负相关。
? KCNN3(书上看看吧)
2) 糖尿病(diabetes mellitus)
临床表现
? 胰岛素依赖型糖尿病(IDIM)
也称为I型糖尿病或青少年型糖尿病,常在青少年期就发病,起病急、症状重且易发生酮症酸中毒,患者消瘦,必需使用胰岛素控制病情;
? 非胰岛素依赖型糖尿病(NIDIM)
常发生于中年以后,称为II型糖尿病,患者一般都较肥胖,起病缓慢、症状较轻。
与遗传有关的依据:
? 家族史:25-50%
? 近亲结婚
? 遗传度:75%
? 同卵双生:45-96% ,异卵双生:3%
候选基因
核基因
? 胰岛素基因
定位于11p15,3个外显子、2个内含子组成,目前发现5个位点的突变与糖尿病发生有关。
? 胰岛素受体基因
定位于19p13,22个外显子和21个内含子,突变后受体蛋白改变,影响与胰岛素的特异性结合 ? 葡萄糖激酶(GCK)基因
定位于7p,10个外显子,多发生错义突变,GCK结构改变,酶活性降低。GCK的突变可导致一种青少年起病的成年型糖尿病
? HLA多态性
各种HLA等位基因本身并不一定直接导致糖尿病,但可能协同糖尿病的易感基因发挥作用。 线粒体基因
? mtDNA中tRNA基因3243位点A→G突变与NIDDM相关。
3) 支气管哮喘(bronchial asthma)
临床特征
临床特点是发作性伴有哮鸣音的呼吸困难,长期反复发作常并发慢性支气管炎和肺气肿。
18
? 外源性哮喘 刺激因素为抗原性的
? 内源性哮喘 刺激因素非抗原性因素
遗传因素 IgE调节基因
第十六章 遗传与肿瘤发生 Cancer Genetics
1、肿瘤的遗传:
1) 单基因遗传的肿瘤
视网膜母细胞瘤家系 (A:AD遗传家系;B:散发性病例家系)
2) 多基因遗传的肿瘤多基因遗传的肿瘤大多是一些常见的恶性肿瘤,这些肿瘤的发生是遗传因素和环境因素共同作
用的结果。如乳腺癌、胃癌、肺癌、前列腺癌、子宫颈癌等,患者一级亲属的患病率都显著高于群体患病率。
3) 染色体畸变与肿瘤 大多数恶性肿瘤细胞的染色体为非整倍体,而且在同一肿瘤内染色体数目波动的幅度较大。 干系(stemline)
在某种肿瘤内,如果某种细胞系生长占优势或细胞百分数占多数,此细胞系就称为该肿瘤的干系,干系的染色体数目称为众数(model number)
旁系(sideline)
如果一种异常的染色体较多地出现在某种肿瘤的细胞内,就称为标志染色体(marker chromosome)。例如:慢性粒细胞性白血病(CML)中的费城染色体(Philadelphia chromosome)
4) 某些遗传性缺陷或疾病具有易患肿瘤的倾向性 例如:共济失调性毛细血管扩张症、Bloom综合征(多见于东欧
犹太人的后裔。患者身材矮小,对日光敏感,故面部常有微血管扩张性红斑。早年发生的癌症(如白血病、林巴瘤))、着色性干皮病、Fanconi贫血症、Werner Syndrome
2、癌基因(oncogene)能够使细胞发生癌变的基因统称为癌基因。他们原是正常细胞中的一些基因,是细胞生长发育所必需的。一旦这些基因在表达时间、表达部位、表达数量及表达产物结构等方面发生了异常,就可以导致细胞无限增殖并出现恶性转化。可分为:病毒癌基因(viral oncogene,v-onc)、原癌基因(proto-oncogene,pro-onc)、细胞癌基因(cellular oncogene,c-onc)或是按功能分类:生长因子受体、信号传递蛋白类细胞癌基因、生长因子,刺激细胞增生、核内转录因子类细胞癌基因
3、原癌基因的突变与肿瘤发生
1) 点突变:原癌基因中由于单个碱基突变而改变编码蛋白的功能,或使基因激活并出现功能变异。例如:膀胱癌细
胞系的ras癌基因第12位密码子GGC突变为GTC,使甘氨酸变为缬氨酸。
2) 染色体易位:由于染色体断裂与重排导致细胞癌基因在染色体上的位置发生改变,并使其激活及具有恶性转化的
功能。例如:Ph染色体
3) 基因扩增:如在40%的神经母细胞瘤细胞中,N-myc原癌基因被扩增了200倍以上。这种基因扩增被认为可产
生原癌基因的过量表达。
4) 病毒诱导与启动子插入:原癌基因附近一旦被插入一个强大的启动子, 如逆转录病毒基因组中的长末端重复序列
(long terminal repeat sequence,LTR),也可被激活。
4、肿瘤抑制基因(recessive oncogene)肿瘤抑制基因也称抑癌基因或隐性癌基因。
1) RB1基因
2) p53、BBCAJ等是其他一些常见的肿瘤抑制基因
例如:视网膜母细胞瘤的RB1。基因视网膜母细胞瘤是婴儿视网膜发生的恶性肿瘤,发病率约l/20000个活婴,大约40%的视网膜母细胞瘤是遗传性的,子代通过生殖细胞遗传一个突变的RB1基因。
5、二次突变假说(two-hit hypothesis)
二次突变假说认为遗传性视网膜母细胞瘤家族连续传递时,已经携带了一个生殖细胞系的突变,此时若在体细胞(如视网膜细胞)内再发生一次体细胞突变,即产生肿瘤,这种事件较易发生,所以发病年龄较早;而散发性的视网膜母细胞瘤是由于一个细胞内的两次体细胞突变而产生的,发生率较低或不易发生,所以发病年龄一般较晚。
6、基因杂合性丢失与肿瘤发生 杂合性丢失在Wilms瘤等许多肿瘤中均有发现,包括遗传性和散发性肿瘤。
19
02遗传
大题4道:
1)14/21平衡易位(7’)
2)血红蛋白病的分子机制(7')
3)先天性聋哑患者与表妹结婚,育有两正常儿女,求再生一个后代发病的概率(Baye
s法)(5')
4)谈谈你对内因(遗传因素)和外因(环境因素)共同作用对疾病发生这一观点的看
法(6')
noun explaination: 10
static mutation
expressivity
heteroplasmy
mosaic
PKU
Turner's syndrome
spina bifida occulta
semizogate
karyotype
heritability
03级
名解
遗传异质性
系谱
半合子
血友病A
出生缺陷
edward综合症
易患性
生物适合度
LHON
光复活修复
1.为什么X连锁显性遗传女性发病率是男性的一倍,但男性的病情较女性的严重?
2.地中海贫血的分类及机制
3.一对夫妇,女性怀孕40天,做染色体检查正常,男性5号染色体发现有臂间倒位(还有一
些废话忘了),从优生的角度看,此时应该作何种检查?胎儿的核型有哪几种情况?应该 采取什么措施? 4.一对夫妇生下一患儿:皮肤,毛发及视网膜颜色较浅,尿中含有特殊的鼠臭味...父亲是 B型血友病患者,这对夫妇想再生一个孩子,请你给他们做遗传咨询。 20