《天然药物化学实验》课程
课程编号:0741532016
实 验 指 导 书
主撰人 解军波
审核人
天津商业大学 生物技术与食品科学学院
二零零九年十月
前 言
1. 实验总体目标
天然药物是人类防病治病药物的一个重要来源。提取、分离天然药物中具有生理活性的化学成分,是天然药物化学研究的一个重要课题。天然药物化学实验主要是学习天然药物有效成分的提取、分离、纯化、鉴定的方法。通过实验,加深学生对理论知识的深入理解,训练学生的基本操作技能,培养学生分析问题和解决问题的能力,为将来从事相关研究打下坚实的实践基础。
⒉ 适用专业年级 制药工程专业 三年级(第五学期)
⒊ 先修课程 分析化学、有机化学
⒋ 实验课时分配
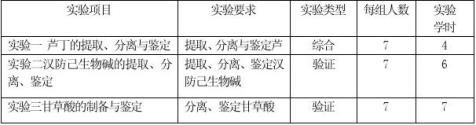
⒌ 实验环境
实验室的标准温度为20℃,实验室内的相对湿度一般应保持在50-70%。 实验室的噪音、防震、防尘、防腐蚀等方面的环境条件应符合在室内开展的检定项目之检定规程。 ⒍ 实验总体要求
提取、分离、纯化、鉴定是贯穿于天然药物化学实验的全过程的基本操作单元。正确掌握和灵活处理各个环节的操作技能是训练学生严格认真的科学态度与良好工作习惯,促进理论与实践结合的一个重要环节。通过本实验让学生掌握、熟悉天然药物化学提取、分离、鉴识技术的基本原理与基本方法,能够进行日常的天然药物研究工作。
⒎ 本实验的重点、难点及教学方法建议
为了保证实验的正常进行和培养学生良好的实验作风及保证实验质量,学生必须遵守下
列要求:
1、实验前必须充分预习实验讲义,明确实验目的、要求,防止实验盲目性,讲求实验质量。
2、实验时要严格按照规范操作进行,仔细观察实验现象,认真记录有关数据。要善于思考,学会运用所学理论知识解释实验现象,研究实验中的问题。
3、重视安全操作,遵守实验室安全守则,使用易燃、易爆、有毒物品时要小心谨慎,避免事故发生。
4、要认真写好实验报告,实验报告要清楚、简练、整洁。
目 录
实验一、芦丁的提取、分离与鉴定..........................................1 实验二、汉防己生物碱的提取、分离、鉴定..................................3 实验三、甘草酸的制备与鉴定..............................................6
实验一 芦丁的提取、分离与鉴定
一、实验目的
1了解黄酮类化合物的一般性质。
2 熟练掌握一些基本技能-回流、抽滤、重结晶等方法。
二、实验内容
利用常规的提取分离方法从槐米中提取分离芦丁,并进行纯度测定。
三、实验要求
以学生自主训练为主的开放模式组织教学,让学生复习常规提取、分离方法的操作及其注意事项。
四、实验准备
复习有机化学中回流、抽滤、重结晶操作的相关步骤和相关事项。
五、实验原理、方法和手段
1. 芦丁:也叫芸香苷(Rutin)
淡黄色针状结晶,熔点:含三分子结晶水物174~178?C;无水物为188?C。 不溶于乙醚、氯仿、石油醚、乙酸乙酯、丙酮等溶剂,易溶于碱液中呈黄色,酸化后复析出,可溶于浓硫酸和浓盐酸,呈棕黄色,加水稀释复析出。
UV:?MaxMeOH(nm) 259,266sh,299sh,359
2. 槲皮素(quercetin)
黄色结晶,熔点:含二分子结晶水物为313~314?C;无水物为316?C。
溶解度:乙醇1:290;无水乙醇1:23(沸时);可溶于甲醇、乙酸乙酯、冰醋酸、吡啶、丙酮等溶剂;不溶于水、乙醚、苯、氯仿、石油醚。
UV:?MaxMeOH(nm) 255,269sh,301sh,370
六、实验条件
恒温水浴锅、微量熔点测定仪、真空干燥箱、真空泵、三用紫外分析仪、层析缸、层析用玻璃毛细管、铺板器、薄层板加热器
微量抽滤瓶、烧杯(500mL、50mL)、500mL圆底烧瓶、冷凝管、500mL分液漏斗 酒精灯、石棉网、铁架台
乙醇、正丁醇、醋酸、盐酸皆为分析纯,AlCl3,芦丁对照品
七、实验步骤
芦丁提取 称取槐米20g,粉碎后,置烧杯中加热水(50?C以上)200mL,煮沸30分钟
1
(注意补充部分蒸馏水),趁热过滤,滤渣再用50 mL?2煮两次,每次10分钟,合并滤液,放置使析出沉淀,然后抽滤,并用少量水洗涤2~3次,得芦丁粗品。
芦丁精制 取芦丁粗品置500mL圆底烧瓶中,加95%乙醇80mL,水浴上温热5分钟,溶解后加活性炭0.1g(先溶解芦丁后再加),接上冷凝管,在水浴上加热,煮沸10分钟,趁热过滤于另一圆底烧瓶中,得黄色澄明溶液,然后回收乙醇,使溶液剩约5~10mL时停止,倾入小烧杯中,以少量95%乙醇洗涤圆底烧瓶,将洗涤液合并于小烧杯中,使结晶尽量析出,然后过滤,得芦丁精品,称重,计算产率。
芦丁熔点的测定 取芦丁精品0.5g,置真空干燥箱中(80?C)干燥30分钟,在微量熔点测定仪上测定熔点。
八、思考题
黄酮类化合物有哪些提取方法?芦丁的提取还有什么方法?
九、实验报告
认真写好实验报告,实验报告要清楚、简练、整洁,数据处理符合分析要求。内容包括:实验名称、目的要求、实验原理、仪器与试药、操作步骤、实验结果、讨论。
十、注意事项及其它说明
(1).所有实验用水均指蒸馏水。
(2)实验中所用易燃有机溶剂,在需要加热时必须用水浴,且注意防火。
(3).槐米的水提液过滤时,必须加适量棉花吸附黏液质,否则将给下一步抽滤造成困难,亦可先倾掉上清液。
2
实验二 汉防己生物碱的提取、分离、鉴定
一、实验目的
1掌握亲脂性生物碱与水溶性生物碱的分离、纯化方法。
2熟悉生物碱的检识和鉴定方法。
3熟悉用硅胶柱层析分离汉防己乙素。
二、实验内容
从粉防己中提取、分离鉴定粉防己碱。
三、实验要求
以学生自主训练为主的开放模式组织教学,让学生熟悉生物碱的提取、分离、鉴别方法的操作及其注意事项。
四、实验准备
预习生物碱的结构、基本理化性质和提取分离方法等相关知识。
五、实验原理、方法和手段
汉防己又名粉防己,为防己科植物粉防己(Stephania tetrandra S.Moder)的干燥根,是一退热镇痛药物,目前该药研究日益广泛。临床上该药除用作治疗神经痛、关节炎、高血压、抗阿米巴原虫外,近来粉防己生物碱的碘甲基或溴甲基化合物已被广泛应用于肌肉松弛剂。汉防己的有效成分是生物碱,总碱含量为1~2%,其中主要的是汉防己甲素、汉防己乙素和少量的水溶性季铵生物碱-轮环藤酚碱和木兰花碱。甲素具有抗风湿及镇痛作用。
1.汉防己甲素(tetrandrine)
本品为无色针状结晶,不溶于水,易溶于甲醇、乙醇、丙酮、氯仿和苯。熔点:217~218?C,有双熔点现象。自丙酮中结晶者105?C左右熔化后加热又固化,至217~218?C复熔。
UV:?MaxMeOH(nm) 280
IR:?maxKBr(cm-1):3050、2800、1605、1580、1500、1235、1215、840
2.汉防己乙素(demethyl tetrandrine fungchinoline)
本品为无色针状结晶,不溶于水和苯,易溶于氯仿,溶于甲醇、乙醇。溶解度与汉防己甲素相似,极性较汉防己甲素稍高,故在苯中溶解度小,而在乙醇溶解度大于汉防己乙素,借此可以将二者分离。汉防己乙素同样具有双熔点现象,熔点因结晶时所用溶剂不同而不同。 丙酮中结晶者:熔点:134~136?C。甲醇中结晶者:熔点:177~179?C。
六、实验条件
1.恒温水浴锅、真空干燥箱、真空泵、三用紫外分析仪、层析缸、层析用玻璃毛细管、铺板器、比重计、薄层板加热器、层析柱
2.微量抽滤瓶、烧杯(500mL、50mL)、500mL圆底烧瓶、冷凝管、500mL分液漏斗、
3
表面皿、三角瓶
3.普通滤纸、层析用硅胶、薄层层析用硅胶G、CMC-Na、无水Na2SO4
4.甲醇、乙醇、丙酮、盐酸、氨水、氯仿皆为分析纯,碘化铋钾试剂
5.汉防己甲素、汉防己乙素对照品
七、实验步骤
(一)总生物碱的提取
取粉防己300g,用95%乙醇回流提取一次(800mL95%乙醇加2 mL浓盐酸回流1小时),四层纱布过滤,合并提取液,减压回收乙醇至小体积,移入蒸发器中水浴浓缩至无醇味,得糖浆状物。
(二)分离
1亲脂性生物碱与水溶性生物碱的分离
在糖浆状物加入3.6%的盐酸约50mL,充分搅拌使生物碱溶解,放置过滤,滤渣用酸水洗涤数次(10mL?2)后弃去。(留酸水液5mL,供鉴定用)合并酸液后移至500mL分液漏斗中,加入80氯仿,再滴加浓氨水调PH为9~10,振摇萃取,分取氯仿层。碱水层再以新氯仿萃取(40mL),合并氯仿液即亲脂性生物碱的提取液;碱水层则为水溶性生物碱提取液。
(三)生物碱的鉴定
薄层层析
硅胶G-CMC-Na板
展开剂:氯仿:丙酮:氨水=5:4:2~3滴
显色剂:碘化铋钾
对照品:汉防己甲素、乙素乙醇溶液(0.1mg/ml)
样品:总防己提取液、酸水液、碱水液
点样量:10微升
八、思考题
1粉防己中含有哪些主要成分?
2糖浆状物中加入3.6%的盐酸的目的何在?
3用1% NaOH萃取的目的何在?
4为什么用水洗至中性?
九、实验报告
认真写好实验报告,实验报告要清楚、简练、整洁,数据处理符合分析要求。内容包括:实验名称、目的要求、实验原理、仪器与试药、操作步骤、实验结果、讨论。
4
实验三 甘草酸的制备与鉴定
一、实验目的
1、掌握高速逆流色谱分离纯化的原理
2、熟悉高速逆流色谱、中压制备色谱的操作方法
二、实验内容
利用高速逆流色谱等方法制备甘草酸,并进行鉴别。
三、实验要求
以学生自主训练为主的开放模式组织教学,让学生熟悉高速逆流色谱的操作及其注意事项。
四、实验准备
复习天然药物化学中的相关知识,预习相关方法和相关事项。
五、实验原理、方法和手段
1、HSCCC利用了一种特殊的流体动力学(单向流体动力学平衡)现象。其原理是基于样品在旋转螺旋管内的互不混溶的两相溶剂间分配不同而获得分离,因而无须任何固体载体或支撑体,能达到在短时间内实现高效分离和制备,并且可以达到几千个理论塔板数。与其他柱色谱相比较,它克服了固定相载体带来的样品吸附、损失、污染和峰形施尾等缺点。
2、甘草酸:本品为白色粉末,有甜味。分子式及分子量:C42H62O16 822.92 。熔点220℃,有特殊甜味, ,溶于热水和热的稀乙醇,不溶于无水乙醇。
六、实验条件
TBE-300A型高速逆流色谱仪系统、中压色谱系统、旋转蒸发器、真空泵、真空干燥箱、熔点测定仪
分液漏斗、烧杯、表面皿
反相硅胶(SP-120-40/60-ODS-B)、乙酸乙酯、甲醇、乙醇、正丁醇、丙酮、盐酸、氨水皆为分析纯,蒸馏水、
甘草药材 甘草酸对照品

5
七、实验步骤
1甘草酸粗提物的制备:
取甘草药材100g,粉碎,过筛(40目),用95﹪乙醇(1:4)超声提取三次,每次45分钟。合并提取液减压抽滤,旋转蒸发仪浓缩得褐色浸膏。在浸膏中加2﹪NaOH溶解,过滤,滤液用2﹪HCl酸化至不再有沉淀生成,过滤,滤渣用冰水洗涤后用真空干燥器在50℃下干燥,得甘草酸粗品。
2高速逆流色谱分离:
2.1供试液的制备:
精密称取甘草酸粗品100mg,溶解于10ml乙酸乙酯-正丁醇-水(5:1.5:5)上层溶液中。
2.2分离纯化:
将供试液注于色谱仪中,收集相应部位,回收溶剂,得纯度较高的甘草酸。
分离条件如下:固定相 乙酸乙酯-正丁醇-水(5:1.5:5)(氨水适量)
流动相 乙酸乙酯-正丁醇-水(5:1.5:5)(氨水适量)
流速 1.5ml/min 转速 1000rpm
温度 25℃ 检测波长 254nm
3中压色谱分离:
3.1 装柱 取反相硅胶500g, 于105℃活化2h后,取出放置室温即可装柱。柱子采用干法填充,填满整个柱体,不留死体积。
3.2取第3步所得甘草酸,取10ml丙酮溶解,上样,泵人中压柱柱头,以甲醇-水(50:
50),25ml/min的流速进行洗脱,254nm波长检测,收集相应部位,真空干燥,得甘草酸纯品。
4测定紫外图谱,鉴定。
八、思考题
1高速逆流色谱选用分离溶剂体系的原则是什么?
2天然产物的纯度测定常用的方法?
九、实验报告
认真写好实验报告,实验报告要清楚、简练、整洁,数据处理符合分析要求。内容包括:实验名称、目的要求、实验原理、仪器与试药、操作步骤、实验结果、讨论。
十、注意事项及其它说明
注意仪器的安全操作
6
第二篇:药物分析实验指导书(11版大纲)
药物分析实验指导书
实验一烟酸原料药的鉴别实验
一、实验目的
1、掌握鉴别烟酸的原理及方法
2、掌握紫外分光光度法鉴别烟酸的方法原理及紫外吸收图谱的解析
3、熟悉紫外分光光度计的操作要点及紫外分光光度法效能指标评价的内容与要求
二、实验原理
1、鉴别反应
(1)烟酸加2,4-二硝基氯苯加热溶化后,生成季铵化合物,再加乙醇制氢氧化钾溶液,即显紫红色,以此鉴别烟酸,反应式为:
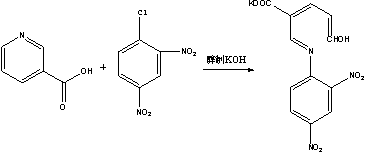
本反应需在无水的条件下进行
(2)烟酸与氢氧化钠发生酸碱中和反应,遇石蕊试纸显中性,遇硫酸铜生成淡蓝色烟酸酮沉淀,以此鉴别烟酸,反应式为:

(3)烟酸加水溶解后,照紫外-可见分光光度法测定,在262nm的波长处有最大吸收,在237nm的波长处有最小吸收,且237nm波长处的吸光度与262nm波长处的吸光度的比值应为0.35~0.39;而烟酰胺也在262nm的波长处有最大吸收,在245nm波长处有最小吸收,在A254nm/A262nm为0.63~0.67。因此可用该方法来区别烟酸和烟酰胺。
三、实验内容与操作
(一)仪器和试剂
1、仪器 紫外分光光度计、配对比色杯一对、试管(25ml,2支)、电炉、药物天平、烧杯(50ml,2只)、容量瓶(100ml、10ml各2只)、移液管(1ml,2只)、乳钵(小号1个,配乳槌)。
2、试剂 烟酸、0.4%氢氧化钠试液(取氢氧化钠0.4g,加水使溶解成100ml,即得)、2,4-二硝基氯苯、乙醇制氢氧化钾试液(取氢氧化钾3.5g,加100ml95%乙醇使溶解,静止后取上清液)、硫酸铜溶液(取硫酸铜12.5g,加水溶解成100ml,即得),蒸馏水、95%乙醇
(二)实验步骤
1、鉴别
(1)取烟酸约4mg,加2,4-二硝基氯苯8mg,研匀,置试管中,缓缓加热溶化后,再加热数秒钟,放冷,加乙醇制氢氧化钾试液3ml,即显紫红色。
(2)取烟酸约50mg,加水20ml溶解后,滴加0.4%氢氧化钠溶液至遇石蕊试纸显中性反应,加硫酸铜试液3ml,即缓缓析出淡蓝色沉淀。
(3)取烟酸,加水溶解并稀释制成每1mL中约含20μg的溶液,照紫外-可见分光光度法(中国药典20##年版附录ⅣA)测定,在262nm的波长处有最大吸收,在237nm的波长处有最小吸收;在237nm波长处的吸光度与262nm波长处的吸光度的比值应为0.35~0.39。
四、思考题
1、计算烟酸的A235nm/A262 nm
五、实验报告书写要求
1、实验目的;2、实验原理;3、主要仪器与试剂;4、实验结果;5、思考题答案。
实验二 阿司匹林原料药的鉴别试验及其特殊杂质的检查
一、实验目的
1、掌握鉴别阿司匹林的原理及方法。
2、掌握阿司匹林中游离水杨酸的检查原理及方法。
3、熟悉阿司匹林中由于合成工艺所带来的其它特殊杂质的检查原理及方法。
二、实验原理
1、鉴别反应
(1)阿司匹林加热水解产生的水杨酸在中性或弱酸性条件下与三氯化铁试液反应,生成紫堇色配位化合物,以此鉴别阿司匹林,反应式为:
COOH COOH






 OCOCH3 + H2O OH + CH3COOH
OCOCH3 + H2O OH + CH3COOH
COOH
 COO-
COO-
6 OH + 4FeCl3 Fe + 12HCl
( O-)2Fe 3
本反应极为灵敏。反应适宜的pH值为4—6,在强酸性溶液中配位化合物分解。
(2)阿司匹林与碳酸氢钠试液加热水解,得水杨酸钠及醋酸钠,加过量稀硫酸酸化后,则生成白色水杨酸沉淀,并发生醋酸的臭气,以此鉴别阿司匹林。反应式为:
COOH COONa
 OCOCH3 + Na2CO3 OH + CH3COONa + CO2
OCOCH3 + Na2CO3 OH + CH3COONa + CO2
COONa COOH
2 OH + H2SO4 2 OH + Na2SO4
 2 CH3COONa + H2SO4 2 CH3COOH + Na2SO4
2 CH3COONa + H2SO4 2 CH3COOH + Na2SO4
沉淀物于100℃—105℃干燥后,熔点为156℃—161℃。
2、特殊杂质检查
(1)溶液的澄清度 本实验系检查碳酸钠试液中不溶物。不溶物杂质有未反应完全的酚类,或水杨酸精制时温度过高,产生脱羧副反应的苯酚,以及合成工艺过程中由副反应生成的醋酸苯酯、水杨酸苯酯和乙酰水杨酸苯酯等。这些杂质均不溶于碳酸钠试液,而阿司匹林可溶解,利用溶解行为的差异,由一定量的阿司匹林在碳酸钠试液中溶解应澄清来加以控制。
(2)游离水杨酸 用高效液相色谱外标法对样品中游离水杨酸的量进行测定
三、实验内容与操作
(一)仪器和试剂
1、仪器 烧杯(50ml, 4只)、电炉、药物天平、配对比浊管(内径15—16mm、平底、具塞、以无色、透明、中性硬质玻璃制成)2支、移液管(5ml 2支)、量瓶(10ml、50ml、 1000ml各2只)。
2、试剂 阿司匹林、水杨酸、三氯化铁试液(取FeCl39g,加水使溶解成100ml,即得)、蒸馏水、碳酸钠试液(取无水Na2CO310.5g,加水使溶解成100ml,即得)、稀硫酸(取硫酸57ml,加水稀释至1000ml,即得)、四氢呋喃、冰醋酸、乙腈(色谱纯)、甲醇。
(二)实验步骤
1、鉴别
(1)取本品约0.1g,置50ml烧杯中,加水10ml,煮沸,放冷,加三氯化铁试液1滴,即显紫堇色。
(2)取本品约0.5g,置50ml烧杯中,加碳酸钠试液10ml,煮沸2分钟后,放冷,加过量的稀硫酸,即析出白色沉淀,并发生醋酸的臭气。
2、检查
(1)溶液的澄清度
【供试品溶液制备】 取本品0.50g,置50ml烧杯中,加温热至约45℃的碳酸钠试液10ml溶解后,制成供试品溶液(供试品溶解后应立即检视)。
操作方法:将规定浓度的供试品溶液在黑色背景下检视
(2)游离水杨酸 取本品0.10g,精密称定,置10ml容量瓶中,加1%冰醋酸的甲醇溶液适量,振摇使溶解,并稀释至刻度,摇匀,作为供试品溶液(临用新制);取水杨酸对照品约10mg,精密称定,置100ml容量瓶中,加1%冰醋酸的甲醇溶液适量使溶解并稀释至刻度,摇匀,精密量取5ml,置50ml容量瓶中,用1%冰醋酸的甲醇溶液稀释至刻度,摇匀,作为对照品溶液。照高效液相色谱法试验。用十八烷基硅烷键合硅胶为填充剂;以乙腈-四氢呋喃-冰醋酸-水(20:5:5:70)为流动相;检测波长为303nm。理论塔板数按水杨酸峰计算不低于5000,阿司匹林与水杨酸的分离度应符合要求。立即精密量取供试品溶液、对照品溶液各20μl,分別注入液相色谱仪,记录色谱图。供试品溶液色谱图如有与水杨酸峰保留一致的色谱峰,按外标法以峰面积计算,不得过0.1%。
四、思考题
1、计算水杨酸的限量。
五、实验报告书写要求
1、实验目的;2、实验原理;3、主要仪器与试剂;4、实验结果;5、思考题答案。
实验三 高效液相色谱法测定阿司匹林原料药的含量
一、实验目的
1、掌握外标法的实验步骤和结果计算方法。
2、熟悉高效液相色谱仪性能检查和色谱参数测定的方法。
3、了解高效液相色谱仪的使用方法。
二、实验原理
(一)阿司匹林的含量测定
1、色谱参数 本实验仅测定下列色谱参数:
(1)理论板数:n=5.54(tR/W1/2)2 理论塔板高度:H=L/n
如果测得的理论板数低于各品种项下规定的最小理论板数,应改变色谱柱的某些条件(如柱长、载体性能、色谱柱充填的优劣等),使理论板数达到要求。
(2)分离度:定量分析时,为便于准确测量,要求定量峰与其他峰之间有较好的分离度。分离度(R)的计算公式为:

式中 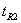 为相邻两峰中后一峰的保留时间;
为相邻两峰中后一峰的保留时间;
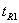 为相邻两峰中前一峰的保留时间;
为相邻两峰中前一峰的保留时间;
W1 及W2为此相邻两峰的峰宽。
除另外有规定外,待测组分与相邻共存物之间的分离度应大于1.5。
(3)拖尾因子 为保证测量精度,特别当采用峰高法测量时,应检查待测峰的拖尾因子(T)是否符合各品种项下的规定,拖尾因子计算公式为
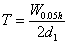
式中, 为5%峰高处的峰宽;
为5%峰高处的峰宽;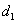 为峰顶点至峰前沿之间的距离。
为峰顶点至峰前沿之间的距离。
除另有规定外,峰高法定量是T应在0.95~1.05之间。
中国药典规定,在采用HPLC法检测药品时,必须对仪器进行系统适用性试验,其试验内容即为理论板数、分离度、拖尾因子以及重复性。
2、高效液相色谱仪的性能指标:本实验仅检查重复性。
重复性:取各品种项下的对照溶液,连续进样5次,除另有规定外,其峰面积测量值的相对标准偏差(RSD)应不大于2.0%。
3、外标法定量分析阿司匹林含量
精密称取对照品和供试品,配制成溶液,分别精密取一定量,注入仪器,记录色谱图,测量对照品溶液和供试品溶液中待测成分的峰面积,按下式计算含量:
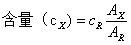
式中 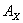 为供试品的峰面积或峰高
为供试品的峰面积或峰高
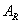 为对照品的峰面积或峰高
为对照品的峰面积或峰高
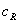 为对照品的浓度
为对照品的浓度
 为供试品的浓度
为供试品的浓度
三、实验内容与操作
(一)仪器和试剂
1、仪器 高效液相色谱仪、ODS柱、万分之一分析天平、量筒 (100ml、 1000ml 各1个)、0.45mm微孔滤膜(油系膜)、量瓶(100 ml,2只)
2、试剂 阿司匹林、四氢呋喃、乙腈(色谱纯)、冰醋酸(AR)、重蒸馏水。
(二)实验步骤
1、阿司匹林含量测定:照高效液相色谱法测定。
色谱条件与系统适用性试验 用十八烷基硅烷键合硅胶为填充剂;以乙腈-四氢呋喃-冰醋酸-水(20:5:5:70)为流动相;检测波长为276nm。理论塔板数按阿司匹林峰计算不低于3000,阿司匹林与水杨酸的分离度应符合要求。
测定法 取阿司匹林原料适量,精密称定,,加1%冰醋酸的甲醇溶液适量,振摇使溶解,并定量稀释制成每1ml中约含0.1mg的溶液,作为供试品溶液;另取阿司匹林对照品同法配制,作为对照品溶液。精密量取供试品溶液、对照品溶液各20μl,分別注入液相色谱仪,记录色谱图。按外标法以峰面积计算,即得。
(三)注意事项
实验中可通过选择适当长度的色谱柱,调整流动相中有机相与水相的比例或流速,使阿司匹林峰、水杨酸峰的分离度达到定量分析的要求。
四、思考题
1、配制供试品溶液时,为什么要使其浓度与对照品溶液的浓度相接近?
2、什么是分离度?如何提高分离度?
五、实验报告书写要求
1、实验目的;2、实验原理(简述);3、主要仪器与试剂;4、实验结果;5、回答思考题。
实验四 中和滴定法测定烟酸的含量
一、实验目的
1、掌握中和滴定法测定烟酸含量的原理及方法
2、掌握中和滴定法测定烟酸含量的操作要点、注意事项与结果计算
3、熟悉中和滴定法的终点确定、效能指标评价的内容与要求
二、实验原理
烟酸为一元强酸性物质,因此,可直接采用强酸的酸碱滴定法测定其含量。具体方法为以酚酞为指示剂,用氢氧化钠标准滴定液滴定烟酸的水溶液,根据消耗的氢氧化钠标准滴定液的用量,计算以C6H5NO2计的烟酸的含量,并将滴定的结果用空白试验校正。
三、实验内容与操作
(一)、仪器与试剂
1、仪器:三角烧瓶(100ml、 12只)、碱式滴定管(25ml、一根)、酸式滴定管(50ml,1根)、量筒(50ml,2只)、万分之一电子天平
2、试剂:0.1mol/L氢氧化钠滴定液(取澄清的氢氧化钠饱和溶液5.6ml,加新沸过的冷水使成1000ml,即得)、水、酚酞指示液(取酚酞1g,加乙醇100ml使溶解,即得)
(二)、实验步骤
1、氢氧化钠滴定液(0.1mol/L)的标定
取在105℃干燥至恒重的基准邻苯二甲酸氢钾约0.6g,精密称定,加新沸过的冷水50mL,振摇,使其尽量溶解;加酚酞指示液2滴,用本液滴定,在接近终点时,应使邻苯二甲酸氢钾完全溶解,滴定至溶液显粉红色。每1mL氢氧化钠滴定液(1mol/L)相当于20.42mg的邻苯二甲酸氢钾。根据本液的消耗量与邻苯二甲酸氢钾的取用量,算出本液的浓度。平行测定3次,3次结果的相对平均偏差不得大于0.1%
注:本滴定液应置聚乙烯塑料瓶中,密封保存;塞中有2孔,孔内各插入玻璃管1支,1管与钠石灰管相连,1管供吸出本液使用。
2、烟酸的含量测定
(1)、实验方法:取本品0.3g,精密称定,加50mL新沸过的冷水溶解后,加3滴酚酞指示液,用氢氧化钠标准滴定液(0.1mol/L)滴定至粉红色,记录消耗氢氧化钠滴定液的体积数(mL),每1 mL氢氧化钠滴定液(0.1mol/L)相当于12.31mg的烟酸(C6H5NO2),即得。
(2)结果计算
烟酸(以C6H5NO2计)的质量分数w1,数值以%表示,按下式计算:
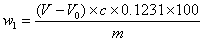
其中:V--消耗氢氧化钠标准滴定液的体积数,单位为mL;
V0--空白实验消耗氢氧化钠标准滴定液的体积数,单位为mL;
c--氢氧化钠标准滴定液实际浓度数值,单位为mol/L;
m--实验室样品的质量数值,单位为g;
注:该实验测得的为烟酸湿品的含量,测得烟酸干燥失重的结果后可将该结果换算为干燥品的含量。
本实验平行操作2次,允许相对差在0.3%以内。
四、思考题
1、什么叫中和滴定突跃范围。
2、根据什么选择酸碱指示剂?举例说明。
3、如何计算氢氧化钠标准滴定液的浓度
4、如果标定氢氧化钠滴定液的基准未烘干,将使标准溶液浓度的标定结果偏高还是偏低?
五、实验报告书写要求
1、实验目的;2、实验原理;3、主要仪器与试剂;4、实验结果;5、思考题答案。