430139生物工程(Biotechnology Engineering)
全日制工程硕士专业学位
研究生培养方案
培养单位: 生命科学学院(204)
药学院(306)
一、培养目标
培养掌握生物工程领域坚实的基础理论和宽广的专业知识,具有较强的解决实际问题的能力,能够独立承担专业技术或管理工作,具有良好的职业素养的高层次应用型专门人才,具体要求为:
1.拥护党的基本路线和方针政策,热爱祖国,遵纪守法,具有良好的职业道德和敬业精神,具有科学严谨和求真务实的学习态度和工作作风,身心健康。
2.掌握本领域的基础理论、先进技术方法和手段,在领域的某一方向具有独立从事工程设计、工程实施,工程研究、工程开发、工程管理等能力。
3.掌握一门外国语。
二、领域简介
主要涵盖:植物生物技术、动物生物技术、生物技术医药、生态技术、发酵工程、基因工程、蛋白质工程、细胞工程、生化工程等。
三、招生对象与学习年限
具有国民教育序列大学本科学历 ( 或本科同等学力 ) 人员。
采用全日制学习方式,学习年限一般为2年。
四、培养方式
采用课程学习、实践教学和学位论文相结合的培养方式。
课程设置体现厚基础理论、重实际应用、博前沿知识,着重突出专业实践类课程和工程实践类课程。实践教学是全日制工程硕士研究生培养中的重要环节,鼓励工程硕士研究生到企业实习,可采用集中实践与分段实践相结合的方式。工程硕士研究生在学期间,必须保证不少于半年的实践教学,应届本科毕业生的实践教学时间原则上不少于1年。
五、课程设置
攻读本领域工程硕士学位的研究生,应获得总学分不少于34学分:
(一)学位课程不低于20学分,包括公共课10学分、专业必修课10学分;
(二)选修课程不低于8学分;
(三)专业实践6学分。
另外,须完成开题报告、论文中期报告两个必修环节。具体课程设置及学分要求见附表。
六、实践环节
在学期间必须保证不少于半年的专业实践,可采用集中实践与分段实践相结合的方式。可在学院已建立的联合培养基地进行实践,也可以结合工程项目到用户单位实践。根据工程项目的要求,需要在本校实践的研究生,需由导师提出书面申请,学院审核批准,并报研究生院审查备案。
研究生须在第1学期期末确定论文导师后,在导师指导下制定并提交实践计划;专业实践一般应安排在第2、3学期期间,并按预先计划的方案逐步完成;每个专业实践环节完成后,研究生须做自我鉴定,并由指导该环节的指导人(可以是校内外导师、也可以是实践部门的专家)作出评定;专业实践完成后,研究生须撰写实践总结报告。研究生一般应结合专业实践确定学位论文的选题。实践表现、总结报告经导师组成的评定小组评审通过后,研究生可获得相应的学分,方可申请进行学位论文答辩。
《武汉大学专业学位研究生实践手册》是研究生院专门制定的重要材料,专业学位研究生每人一册,用以实时并详细记载其专业实践各环节的实施情况与评定,请务必在完成后连同实践总结报告一起存入研究生的学位档案。
七、学位论文
论文选题应来源于工程实际或具有明确的工程技术背景,可以是新技术、新工艺、新设备、新材料、新产品的研制与开发。论文的内容可以是:工程设计与研究、技术研究或技术改造方案研究、工程软件或应用软件开发、工程管理等。
论文工作须在导师指导下独立完成。实行双导师制,其中一位导师来自校内,另一位导师为来自企业与本领域相关的专家。
论文撰写完成后除经导师写出详细的评阅意见外,还应有2位(其中至少一位来自校外)本领域或相近领域的专家评阅。论文评审应重点审核:论文作者综合运用科学理论、方法和技术手段解决工程技术问题的能力;论文工作的技术难度和工作量;其解决工程技术问题的新思想、新方法和新进展;其新工艺、新技术和新设计的先进性和实用性;其创造的经济效益和社会效益等方面。
攻读全日制工程硕士研究生完成培养方案中规定的所有环节,获得规定的学分,成绩合格,方可申请论文答辩。答辩委员会应由5位(其中至少一位来自校外)与本领域相关的专家组成。
通过论文答辩者,经校学位评定委员会审核通过,可授予工程硕士专业学位,同时获得硕士研究生毕业证书。
第二篇:Genetic Engineering and Cloning Focus on Animal Biotechnology
Chapter 4
Genetic Engineering and Cloning:Focus on Animal Biotechnology
Mariana Ianello Giassetti*,
Fernanda Sevciuc Maria*,
Mayra Elena Ortiz D’ávila Assump??o and
José Ant?nio Visintin
Additional information is available at the end of the chapter/10.5772/56071
1. Introduction
1.1. What is genetic engineering?
Over the last 35 years the term genetic engineering has been commonly used not only in sciencebut also in others parts of society. Nowadays this name is often associated by the media forensictechniques to solve crimes, paternity, medical diagnosis and, gene mapping and sequencing.The popularization of genetic engineering is consequence of its wide use in laboratories aroundthe world and, developing of modern and efficient techniques. The genetic engineering, oftenused with trivia, involves sophisticated techniques of gene manipulation, cloning andmodification. Many authors consider this term as synonymous as genetic modification, wherea synthetic gene or foreign DNA is inserted into an organism of interest. Organism that receivesthis recombinant DNA is considered as genetically modified (GMO). Its production aresummarized in simplified form in five steps: 1) Isolation of interested gene, 2) Construction,gene of interested is joined with promoters (location and control the level of expression),terminator (indicates end of the gene) and expression marker (identify the gene expression),3) transformation (when the recombinant DNA is inserted into the host organism), 4) Selection(selection of those organisms that express the markers), 5) Insertion verification of recombinantDNA and its expression [1].
1.2. How to apply genetic engineering in our everyday
One of the main firstlings of genetic engineering is that genetic information is organized in theform of genes formed by DNA, which across some biotechnologies can be manipulated to be
? 2013 Giassetti* et al.; licensee InTech. This is an open access article distributed under the terms of the
Creative Commons Attribution License (http://creativecommons.org/licenses/by/3.0), which permitsunrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
64Genetic Engineering
applied in various fields of science. Currently, genetic engineering is widely used at variousbranches of medicine to produce vaccines, monoclonal antibodies, animals that can be usedas models for diseases or to be used as organ donors (such as pigs). Another function of geneticengineering is gene therapy which aims to restore correct gene expression in cells that have adefective form. In the industry, genetic engineering has been extensively used for the produc‐tion bioreactor able to express proteins and enzymes with high functional activity. Already inagriculture, genetic engineering is being very controversial because it tends to producegenetically modified foods resistant to pests, diseases and herbicides.
1.3. Concept is already old
However, all the knowledge obtained in the present day was only possible by discoveries ofGregor Mendel, considered the father of genetics. The results obtained in 1865 by the Austrianmonk generated genetics studies related to heritability and variation. The term formerly called"element" by Mendel was later termed "genes" by Wilhelm Johanssen in 1909. Sutton andBoveri (1902) have proposed that these genes were grouped in the form of chromosomes, whichin turn constitute the genetic material of eukaryotes. In 1953, James Watson and Francis Crickunraveled the structure of DNA as double helix, creating a period of intense scientific activitythat culminated in 1966 with the establishment of the complete genetic code.
Major new discoveries were made in 1967 when DNA ligase was isolated that has the abilityto join DNA fragments. The first restriction endonuclease enzyme was isolated in 1970 and itfunctions as a scissors cutting a specific DNA sequence. These discoveries allowed thedevelopment of the first recombinant DNA molecule, which was first described in 1972. In1973 restriction enzymes (scissors) and DNA ligase (adhesive) were used to join a DNAfragment in plasmid pSC101, which is a circular extrachromosomal bacterial DNA. Thus, E.coli was transformed with the recombinant plasmid and it was replicated, generating multiplecopies of the same recombinant DNA. The experiments conducted in 1972 and 1973 werecrucial to the establishment of new genetics and genetic engineering.
1.4. Genome: Structure, organization and function
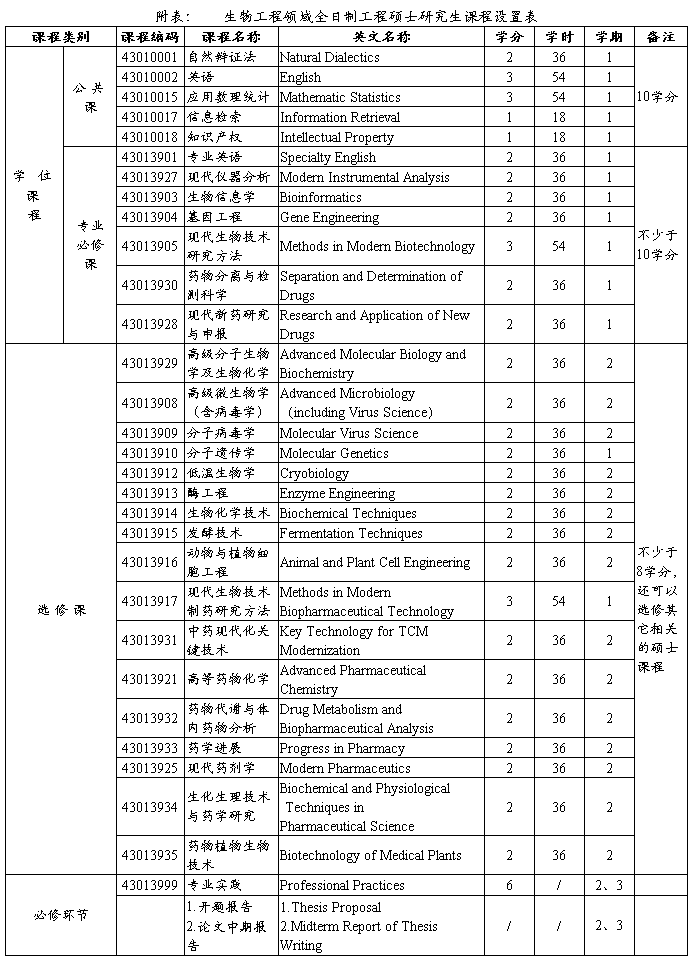
Genome is considered long chains of nucleic acid that contains the information necessary toform an organism [2], consisting of small subunits called nucleic bases that are inheritable.Thus, the genome contains a complete set of features that are inheritable. The genome canbe divided functionally into sets of base sequences, called genes. Each gene is responsiblefor coding a protein, and alternative forms called alleles. A linear chain gene is namedchromosome and each gene assumes a specific place, locus. Therefore, the modern view ofgenetics genome is a complete set of chromosomes for each individual. According to thecentral dogma (Figure 1), each gene sequence encodes another sequence of nitrogenous basesof single stranded RNA. The RNA sequence, complementary to a genomic DNA, will encodeamino acids that form the protein. As previously mentioned, each gene relates with expressionof one protein and for that each codon (the sequence of 3 nitrogenous bases of DNA) representonly one amino acid, but each amino acid can be represented by more than one codon.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607165
Figure 1. Central Dogma, gene codes a RNA sequence that is complementary of DNA and it encodes a protein.
1.5. DNA and RNA structure
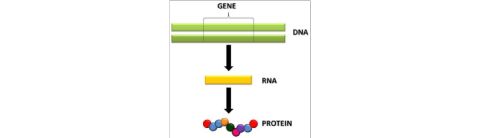
The DNA is considered as genetic material of bacteria, viruses and eukaryotic cells havinga basic structure the nucleotide, which is formed by a nitrogenous base (purine ring orpyrimidine), sugar and phosphate. In 1953, Watson and Crick proposed that DNA is a doublepolynucleotide chain organized as a double helix. In this model, the double helix was linkedby hydrogen bounding between nitrogenous bases. The base is linked to the 1-position by apentose glycosidic bond from N7 of pyrimidines or N9 of purine. The nuclear acid is namedby the type of sugar. DNA has 2`-deoxyribose, whereas RNA has ribose. The sugar in RNAhas an OH group in a 2` position of pentose ring. A nucleic acid is a long chain of nucleoti‐des and the sugar can be linked in 3′or 5′ position to the phosphate group and the back‐bone of chain consist in a repeated sequence of sugar (pentose) and phosphate residues. Onepentose ring is connected at 5`position to a forward pentose that is linked by the 3` posi‐tion via phosphate residues; in this way, the sugar-phosphate backbone is 5′-3` phosphodiest‐er linkages (Figure 2).
Nucleic acid contains 4 types of base, 2 purines (adenine (A) and guanine (G), which are presentin DNA and RNA) and two pyrimidines (cytosine (C) and thymine (T) for DNA and for RNAuracil (U) instead of thymine). Therefore, DNA contains A, G, C, T and RNA contains A, G,C and U. Other important discover were that the G bounded specifically with C, and T/U withA; these named base pairing (complementary), and that the chains had apposite directions(antiparallel).
2. Genetic engineering: Timeline
The chronological order of main events of genetic engineering and cloning are described above.1866 - Gregor Mendel proposed the law of independent, of segregation and basic principlesof heredity; principles that created a new science called genetic.
66Genetic Engineering
Figure 2. Polynucleotide chain, 5′-3′sugar phosphate linkages (backbone) and structure of nucleotide subunit -Adapted from Lewin, B (2004)[2]
1900 - Mendel′s principles were rediscovered by Hugo de Vries, Carl Correns and Eric vonTschermak
1908 - Chromosome Theory of Heredity was proposed by Thomas Hunt Morgan
1944 - Was established that DNA contains the heredity material.
1946 - First electronic digital computer was created
1952 - The first cloned animal (Northern Leopard Frog)
1953 - Watson and Crick described DNA structure and proposed the double helix model.
1955 - Protein sequencing method was established by Frederick Sanger and insulin wassequenced
1965 - Atlas of protein sequences was created
1966 - Genetic code was cracked
1970 - Algorithm for DNA sequence was created
1972 - Establishment of DNA recombinant technology by Stanley Cohen, Herbert Boyer andPaul Berg
1973 - The first recombinant DNA organism was created
1976 - The first genetic engineering company is founded.
1977 - DNA sequencing method was established
1980 - Was done the first molecular mapping of a human chromosome
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607167
1982 - GeneBank started to be public
1983 - Mullis developed PCR (Polymerase chain reaction)
1984 - "Genetic Fingerprinting" techniques was developed and human genome sequencingstarted
1986 - National Center for Biotechnology was developed in USA and automatic machine forDNA sequencing was created
1990 - Dolly, the first cloned animal, was born and blast program was created
1995 - First complete bacterial genome was sequenced
1997 - E. coli complete genome sequence was published
1999 - Complete sequence of human chromosome 22 was published
2000 - Drosophila genome was sequenced and first holy genome from plant was published2002 - Mouse genome sequence was published
2003 - Human genome sequence was published
2004 - Chimpanzee genome sequence was published
3. Cutting and pasting the DNA
3.1. Discovering restriction endonuclease and a Nobel Price in 1978.
Molecular biology and genetic were innovated in middle of 70th decade the discover ofrestriction endonuclease by W Arber, D Nathans e H Smith that wan the Nobel price in 1978.When phage λ attacks an E. coli strain B a specific restriction endonuclease (EcoB) cuts just theDNA from phage λ and infections is blocked. E.coli methylates its own DNA by action of DNAmethylase to protect this DNA from itself enzyme. Restriction endonuclease recognizes shortsequences of duplex DNA as cleavage target and the enzyme cuts this point of DNA everytime this target sequence occurs. When the DNA molecule is cleaved by restriction endonu‐clease DNA fragments are produced. Analyzing restriction fragments is possible to generatea map of the original DNA molecule (restriction map, a linear sequence of DNA separated indefined fragment size) [1, 2]
3.2. Types of restriction endonuclease enzyme: Nature, structure, application, recognitionsite of action and nomenclature
Restriction endonuclease are classified in types I, II and III by sequence specificity, nature ofrestriction and structural differences (table 1). Types I and III have a restrict use in molecularbiology and genetic engineering but the type II is largest used because it cleaves the DNA aspecific recognition sequence, separate methylation, no additional energy requirement isnecessar, high precision and do not match actions. Type II restriction endonuclease are
68Genetic Engineering
classified by the size of recognition sequence such as tetracutter, hexacutter or octacutter (4, 6and 8 base paired respectively) [3].; and generally that sequences are palindromic (nitrogenousbases sequence read the same backwards and forwards). Restriction enzymes also could beclassified as neoschizomers (recognize the same sequence) and isoschizomers (recognize andcleave in the same location).
Type I
Enzyme structure
Complex of three subunitswith independentrecognition endonucleaseand methylase function
Type II
Separate monomericenzymes for endonucleaseand methylase action, bothrecognize the same targetsequence
Requirement for activationATP and Mg2+
S-Adenosyl methionine
Mg2+
ATP and Mg2+S-Adenosyl methionineEnhance activity
Recognition site
Double-stranded DNA
Generally palindromicsequence of Double-stranded DNA
Nature of restriction
Cleaves the DNA at a randomCleaves the DNA at a specificCleaves the DNA about 25pbsequence at one Kb away ofsequence near or at therecognition site
recognition site
downstream of the
recognition site at a randomsequence
Table 1. Properties of restriction endonucleases – Adapted from Satya, P[3] (2007)
Single-stranded DNA
Type III
Separated dimeric enzymesfor endonuclease andmethylase with one commonsubunit
The nomenclature of restriction endonuclease is derivate from the species that it was isolated(Ex. ECORI, from Escherichia coli Ry13); First two letters from enzyme name identify the speciesand the third identify the different strains from the same organism (Table 2). The numberclassifies the different enzymes from the same organism and strains in chronological order ofdiscover (Ex. Hind III, is the third RE isolated from Haemophilus influenza). Restriction endo‐nuclease cut the DNA in two different ways: blunt end (two DNA strands are cleaved at thesame position) or sticky end (the enzyme cut each DNA strand at different position, generallytwo until four nucleotides apart). So in the sticky, DNA fragments have short single-strandedoverhangs at each end. ) [1-4]
3.3. Linking of DNA fragments: DNA ligase
Restriction endonuclease type II cuts the double-stranded DNA in specific target sequence butthis enzyme do not joined back again the DNA fragments, this is essential to create a newhybrid DNA. Joining two DNA fragments by 5`→3` phosphodiester bond is an energydependent process (ATP or NAD, depending the kind of enzyme that is being used). DNAligase is a specific enzyme that is responsible to join DNA fragments spending and two blunt
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/56071
69
EnzymeAluI
Recognition sequence5′-AGCT-3′3′-TCGA-5′
Type of endsBlunt
End sequences5′-AG ? CT-3′3′-TC ? GA-5′
Sau3AI5′-GATC-3′3′-CTAG-5′
Sticky, 5′ overhang5′- ? GATC-3′3′-CTAG ? -5′
HinfI5′-GANTC-3′3′-CTNAG-5′
Sticky, 5′ overhang5′-G ? ANTC-3′3′-CTNA ? G-5′
BamHI5′-GGATCC-3′3′-CCTAGG-5′
Sticky, 5′ overhang5′-G ? GATCC-3′3′-CCTAG ? G-5′
BsrBI5′-CCGCTC-3′3′-GGCGAG-5′
Blunt5′- ? NNNCCGCTC-3′3′- ? NNNGGCGAG-5′
EcoRI5′-GAATTC-3′3′-CTTAAG-5′
Sticky, 5′ overhang5′-G ? AATTC-3′3′-CTTAA ? G-5′
PstI5′-CTGCAG-3′3′-GACGTC-5′
Sticky, 3′ overhang5′-CTGCA ? G-3′3′-G ? ACGTC-5′
NotI5′-GCGGCCGC-3′3′-CGCCGGCG-5′
Sticky, 5′ overhang5′-GC ? GGCCGC-3′3′-CGCCGG ? CG-5′
gglI5′-GCCNNNNNGGC-3′3′-CGGNNNNNCCG-5′
Sticky, 3′ overhang5′-GCCNNNN ? NGGC-3′3′-CGGN ? NNNNCCG-5′
*N = any nucleotide._
*Note that most, but not all, recognition sequences have inverted symmetry: when read in the 5′→3′ direction, thesequence is the same in both strands.
Table 2. Same restriction endonuclease used in genetic engineering - Adapted from Brown, TA (2002)[4]
ends can be joined easily spending two ATPs molecules and this blunt end is very popular ingenetic engineering. However, the efficiency of this process is very low because the DNA ligasejust joins adjacent DNA fragments and it cannot bring the DNA end nearby. Action of enzymeto catalyze the reaction is a random process that depends of vicinity of DNA fragments insolution. Joining DNA fragments with blunt ends is generally used to short oligonucleotidesbecause concentration of free ends and enzyme are high, increasing the efficiency of process.Presence of sticky ends increase process efficiency because complementary ends come togetherby a random diffusion event in the solution and transient base pair might form between thetwo complementary strand. This ligation is not very stable but may persist for enough time tojoin DNA fragments by DNA ligase catalysis and synthesis of phosphodiester bonds [4].
70Genetic Engineering
The greater efficiency of sticky-end ligation stimulated the creation of new methods, suchlinkers or adaptors. They are short double-strand molecules that cover the blunt-end and inserta recognition sequence for a restriction endonuclease to create a sticky-end. The linkers needto be digest by a restriction endonuclease to have a stick-end but the adaptor is a final sequence,digestion is not necessary and fragments can be direct joined by DNA ligase.
4. DNA cloning
In modern molecular biology the ability to manipulate DNA molecules by restriction endo‐nuclease and DNA ligase is named by DNA cloning and, a recombinant DNA can be con‐structed. However, a single copy of recombinant DNA is not enough. Replication machineryof one organism generally is used to increase the number of copies. The DNA is inserted in theorganism for a propagation or transfer. Generally, the vector has autonomic replication systemthat is independent of the cell cycle, increasing the number of copies. Majority systems of DNAcloning use bacterial as a host and common plasmid vector is classified in low copy number(<10) or high copy number (>20). To select recombinant cell some parameters need to bepresent: have restriction sites in which de exogenous DNA is inserted (just one site for eachrestriction endonuclease) and vector needs to have a marker gene multicloning sites (one sitefor several restriction endonuclease) makes the vector more useful [3, 4].
5. Isolation, sequencing and synthesis DNA
The transgenic animal technology involves in first place, the isolation or artificially synthesisof a gene, which will be molecular manipulated and used for transformation leading to thetransgenic production. The need of knowledge involving this target gene can be overcome byits sequencing, conducting to the understanding of its structure. The main of this subject isbriefly described the mechanisms involved in isolation, sequencing and synthesis of a gene.
5.1. Isolation of genes
The first gene isolation was reported in 1969. Two specialized transducing phages, bacterio‐phages ? and Φ80, which carry the lac operon of Escherichia coli was inserted in reverseorientation into their DNA, being used as a source of complementary sequences to preparepure lac operon duplex [5]. This method besides being resourceful work did not have generalapplicability.
Now a day, several methods are in progress for isolation of a gene. A most traditional methodused largely in research is the construction of a genomic or complementary DNA (cDNA)library. A genomic library represents the total DNA of a cell including the coding and non-coding sequences cloned on a vector and a cDNA library is a combination of cloned fragmentsfrom the mRNA inserts into a collection of host cells, creating in both cases, a portion oforganism transcriptome.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607171
To produce a genomic library after the extraction of genomic DNA, these molecules aredigested into fragments of reasonable size by restrictions endonucleases and then inserted intoa cloning vector generating a population of chimeric vector molecules.
On the other hand, to create a cDNA library is necessary first, to produce a cDNA, which canbe obtained from a mature mRNA isolate from a tissue or cells actively synthetizing proteins.The extraction of mRNA is easy due to poly-A tail present in eukaryotic mRNAs. Than theextracted mRNA is used for copying it into cDNA using the reverse transcriptase enzyme,method that create a single strand cDNA, which is converted in a double strand cDNA withDNA polymerase, coiling and nucleases. This cDNA are cloned into a bacterial plasmid, whichis transformed into bacterial competent cells, amplified and selected.
Once a genomic or a cDNA library is available, they can be used for the identification andisolation of a gene sequence.
There are many commercial kits to create a genomic or cDNA library. Normally, the genomiclibrary is created with lambda or cosmid vectors while a cDNA library is produced withplasmid vectors (more information see item 5). These kits usually try to improve the classicallaborious techniques, enabling rapid construct of the libraries and ensuring generating of full-length clones.
The isolation of a gene using a genomic or cDNA library can be done by colony hybridization.In this technique the fragments containing a gene or parts of it can be identified by the use ofDNA probes, which can be tagged or labeled with a molecular marker of either radioactive orfluorescent molecules. The commonly used markers are phosphorus 32 and digoxigenin, anon-radioactive, antibody-based marker.
The DNA of bacteria carrying the chimeric vectors is fixed on the filter, which is hybridizedwith the labeled probe carrying a sequence related to the gene to be isolated. The coloniescarrying moderate to high similarity to the desired sequence are detected by visualizing thehybridized probe via autoradiography or other imaging techniques. In this way, the originalchimeric vectors carrying the target gene sequence can be recovered from original coloniesand used for advance researches.
If the library available were in the form of phage particles, instead of colonies are plaques thatcan be hybridized in the same way described above for colonies. This method of identificationand isolation of genes are called plaques hybridization.
5.2. Isolation of genes related to a protein
To identify a gene related to a protein the inverse pathway (from protein to DNA) should besimulated. For start is necessary to have the protein product in a pure form. To purify a proteinseveral methods typically used are in a series of steps. Each step of protein purification usuallyresults in some degree of product loss, so, an ideal strategy is one in which the highest levelof purification is reached in the fewer steps. The properties of the protein product like size,charge and solubility; determines the selection of which steps to use. These steps can beprecipitation and differential solubilization; ultracentrifugation or chromatographic methods.
72Genetic Engineering
Thus, having the protein product is possible to produce antibodies probes for this protein byimmunizing animals. This production require reliance upon animals immune system to levyresponses that result in biosynthesis of antibodies against the inject molecule. Antigens mustbe prepared and delivered in a form and manner that maximizes production of a specificimmune response by the animal.
These antibodies probes can be used to precipitation of polysomes engaged in synthesizingthe target protein leading to the achievement of the mRNA coded for this protein. This methodcombined with immunoadsorbent techniques brings the possibility of application at lessabundant proteins expression [6]. Then the mRNA are isolated and purified from the polysomefraction, being after used for synthesizing cDNA for a cDNA library preparation, describedabove.
Thereby, to identify the specific cDNA clone for the target protein immunological andelectrophoretic analysis methods are used, screening a complete or partial genomic library [10].
5.3. DNA Sequencing
The basic concept of DNA sequencing is the mechanism involved in determining the order ofnucleotides bases (adenine, guanine, cytosine and thymine) in a strand of DNA. F. Sanger andcoworkers reported the first DNA sequencing, which was genome of DNA ΦX174 virus. Thus,at that moment, two methods of DNA sequencing were developed: one proposed by A. Maxamand W. Gilbert, known as chemical method of DNA sequencing, and the other developed byF. Sanger, S. Nicklen and A. R. Coulson known as chain termination method.
The chemical method of DNA sequencing consists in determines the nucleotide sequence of aterminally labeled DNA molecule by breaking it at adenosine, guanine, cytosine and thyminewith chemical agents. Partial cleavage at each base produces a nested set of radioactivefragments extending from the labeled end to each of the positions of the base. The autoradio‐graph of a gel produced from four different chemical cleavages, shows a pattern of bands fromwhich the sequences are read directly [7].
The chain termination method depends on DNA replication and termination of replication atspecific sequences. For that, Sanger’s technique is based on an enzymatic synthesis from asingle-stranded DNA template with chain termination on DNA polymerase, using dideoxy‐nucleotides (ddNTPs). The principle of this method relies on the dideoxynucleotide lacking a3’OH group, which is required for extension of the sugar phosphate backbone. Thus, DNApolymerases cannot extend the template copy chain beyond the incorporated ddNTP [3, 8].Both methods rely on four-lane high-resolution polyacrylamide gel electrophoresis to separatethe labeled fragment and allow the base sequence to be read in a staggered ladder-like fashion.Sanger sequencing was technically easier and faster, becoming the main basis of DNAsequencing, being modified and automated to aid large scale sequence procedure [3, 8, 9].
5.3.1. Automatic sequencing
An automatic sequencing is an improvement of Sanger sequencing, through the use of differentfluorescent dyes incorporated into DNA extension products primers or terminator. The use of
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607173
different fluorophores in the four based (A, C, G and T) specific extension reactions means thatall reactions can be loaded in a single lane. For each base one color are used, emitting a differentwavelength when excited. Throughout electrophoresis, the fluorescence signs are detected andrecorded [10, 11].
The classic electrophoresis methods used in automated sequencing are slab gel sequencingsystem or capillary sequence gel system, both described below.
5.3.2. Slab gel sequencing systems
The slab gel sequencing system consists of using ultrathin slab gels, about 75μm, and comprisesrunning of at least 96 lanes per gel. By this instrument, fluorescent-labeled fragments wereloaded to the top of vertical gel and electric filed was applied, as the negatively charged DNAfragments migrated through the gel they were sized and fractionated by the polyacrylamidegel. The fragments were automatically excited with a scanning argon laser and detected by acamera [12].
The loading of sequencing gels samples can be done manually or automatically. The automa‐tion consists in the use of a plexiglass block with wells in same distance from each other as thecomb teeth cut in a porous membrane used as a comb for drawing samples by capillary action.The loading of samples automation achieve up to 480 samples per gel [9].
5.3.3. Capillary sequence gel systems
Alternatively, the capillary sequence gel system instead of continuous polyacrylamide gelslabs, DNA is sent through a set of 96 capillary tubes filled with polymerized gel [3, 9].
In this system fused silica capillaries of 50-100 μm in diameter and 30-80 cm in length, heatresistant, are filled with a separation matrix consisting of a gel and electrode buffer. Solutionphase DNA molecule are injected into the capillary either by pressure or electrokinetic injectionand separated inside the capillary according to their size under high voltage conditions. Themolecules are detected using UV light absorption or laser induced fluorescent detection at theend of the capillary [3, 12].
5.3.4. Direct sequencing by PCR
PCR has relieved much of the experimental toil of molecular biology improving the proce‐dure’s sensibility and facilitating the rapid cloning and sequencing of large numbers of samples[13]. The amplification of target DNA by PCR followed by direct sequencing of amplified DNAhas emerged as a powerful strategy for rapid molecular genetics analysis bypassing the timeconsuming cloning steps and generating accurate DNA sequence information from smallquantities of precious biological samples [14].
The direct PCR sequencing involves two steps 1- generation of sequencing templates throughPCR and 2- sequencing of PCR products using thermolabile or thermostable DNA polymerases[15].
74Genetic Engineering
Some enzymes as Taq polymerase are thermostable and can be used in automated sequencingreactions such as cycle sequencing. Others, such as Klenow polymerase and reverse transcrip‐tase are thermal instable, being able to both direct sequencing by PCR products and clonedtemplate, although cannot be used in cycle sequencing. Another enzyme, Sequenase, has alsobeen used effectively in both radioactive and fluorescence cycle sequencing [8].
One sequencing strategy of form any PCR-amplified DNA template are the sequenaseapproach. First, the PCR-amplified DNA is denatured to single strands, annealing thesequencing primer to complementary sequence on one of the template strands. Then, theannealed primer is extended by DNA polymerase by 20-80 nucleotides, incorporating multipleradioactive labels into the newly synthesized DNA, under non-optimal reactions conditions,retaining the enzyme functionality low, for the synthesis of only short stretches DNA. After,the labeled DNA chains are extended and terminated by incorporation of ddNMPs [14].
On the other hand, cycle sequencing strategies can be used for PCR-amplified DNA. Thesemethods generate high-intensity sequence ladders due to the advantage of automated cyclingcapability of thermal cyclers. First, the PCR-amplified DNA is denatured to single strands, andthen it is annealed of a 32p-labeled sequencing primer. After, it is extended and chain-terminated by a thermostable DNA polymerase and denatured in the next sequencing cycle.This step releases the template strand for another round of priming reactions while accumu‐lates chain-terminated products in each cycle. These steps are repeated 20-40 cycles to amplifythe chain-terminated products in a linear fashion [14].
5.3.5. DNA sequencing by microarray
A DNA microarray technology brings the possibility of large scale sequence analyses bygenerating miniaturized arrays of densely packed oligonucleotide probes [9, 16].
The word microarray has been derived from the Greek word mikro (small) and the French wordarrayer (arranged). This technology can be described as an ordered array of microscopicelements on a planar surface that allows the specific binding of genes or gene products [17, 18].The DNA sequencing by microarray uses a set of oligonucleotide probes to examine forcomplementary sequences on a target strand of DNA. Briefly, after cleavage DNA segmentsare hybridized to the definitely arranged probes on a gene chip, the detection is made with alight driven. Then, to reconstruct the target DNA sequence, the hybridization pattern is used.To analyze the data and determinate the DNA sequence specific software are used [3, 16].The array elements react specifically with labeled mixtures, producing signals that reveal theidentity and concentration of each labeled species in solution. These attributes provideminiature biological assays that allow the exploration of any organism on a genomic scale [17].The array technology has been widely used in functional genomics experiments designed tostudy the functions and interactions of genes within the context of the overall genome distinctplant and animal species. To sequence a DNA fragment by microarray a series of laboratoryprocedures are involved, from RNA extraction, reverse transcription and tagging fluorescenthybridization to the end, which invariably introduce different levels of additional variation
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607175
data. On the other hand, experiments with microarrays are still considerably expensive andlaborious and, as a consequence, are generally conducted with relatively small sample sizes.Thus, the conducting tests on microarrays require careful experimental design and statisticalanalysis of the data [19].
5.3.6. DNA sequencing by MALDI TOF mass spectrometry
The Matrix assisted laser desorption/ionization is very rapid and combined with time-of-flight(MALDI-TOF) became an efficient and less time consuming (range of several microseconds)in acquire DNA sequence information by sensitive discrimination of their molecular masses.The technique consists in embedded the samples to be analyzed in a crystalline structure ofsmall organic compounds (matrix) and deposited on a conductive sample support. Then, thesamples are irradiated with an ultraviolet (UV) laser with a wavelength of 266 or 337nm. Theenergy of the laser causes structural decomposition of the irradiated crystal and generates aparticle cloud from which ions are extracted by an electric field. Following acceleration throughthe electric field, the ions drift through a field-free path and finally reach the detector. Theresults come from the calculation of ion masses by measuring their TOF, which is longer forlarger molecules than for smaller ones. Due to single-charged, nonfragmented ions are mostlyproduced, parent ion masses can be determined from the resulting spectrum without the needfor complex data processing. The masses are accessible as numerical data for direct processingand subsequent analysis [20].
The development of MALDI-TOF for an efficient DNA analyses happens due to needed ofhigh throughput, parallel processing, simplified handling and low-cost techniques. Themethod uses an initial PCR amplification, which, PCR is carried out with a DNA polymerasethat accepts ribonucleoside triphosphates (NTP) substrates. One of the four deoxynucleotidesis replaced by an NTP. Fragments are generated by simple alkali backbone cleavage at the ribo-bases of the PCR products, generating oligonucleotide fragments each terminating with theribonucleotide of the cycled primer extension reaction. Analysis is carried out by MALDI-TOFmass spectrometry. Differences between the unknown sample and a reference sequence aredetermined by changes in the results pattern [21, 22].
Nowadays, with the advent of genome sequencing projects been accomplished, sequences ofDNA can be obtained and compared through electronically databases, than physically fromclone libraries (described above). The available databases include locus information, organismspecies, the whole gene sequence, the reference authors and the status of the sequencing. Themost used resource is the GenBank [23] provided for the National Center for BiotechnologyInformation (NCBI).
5.4. Synthesis
The gene synthesis methods had their main development during 1980s and 1990s. DNA genesynthesis is the process of writing the DNA. As DNA carries the genetic information of anorganism, it could be viewed like a kind of information resource, enabling its reading (se‐quencing, described above) and writing (synthesis).
76Genetic Engineering
The oligonucleotides synthesis can be done rapidly and in high yields with different kinds ofmethods. The gene synthesis, together with the knowledge of full genomes, molecular cloning,and protein expression profiles, improved the biotechnology field, making possible to explorethe whole functionality of an entire complex organism.
5.4.1. Gene synthesis machine
The gene synthesis machine is fully automated instrument, which synthesizes predeterminedpolynucleotide sequence. The principle involved is based on a combination of organicchemistry and molecular biological techniques.
Automatic gene machines, synthesize specific DNA sequences by programming the apparatusfor the desired sequence. Briefly, the chosen sequence is entered in a keyboard and a micro‐processor automatically opens the valve of nucleotide, chemical and solvent reservoir,controlling the whole process [15].
Containers of the four nucleotides (A, T, C and G) and reservoirs for reagent and solventsupports are connected with the synthesizer column. This column is packed with small silicabeads, which provides support for assembly of DNA molecules. The desired sequence issynthesized on the silica beads which are later removed chemically [23].
Commercial services for gene synthesis are available from numerous companies worldwide.This gene synthesis method provides the possibility of creates entire genes without the needof a DNA template.
5.4.2. Gene synthesis from mRNA
The reports of a ribonuclease-sensitive endogenous DNA polymerase activity in particles ofRNA tumor viruses by H.M. Temin and D. Baltimore enable the synthesis of complementaryDNA (cDNA) using mRNA as template [9, 15, 24].
This enzyme, known as reverse transcriptase, are largely used in biotechnology research, andcombined with the polymerase chain reaction create a methodology for DNA synthesis andamplification of the product.
To use the mRNA as a template first is necessary purify this molecule of the cell, or tissue. Thiscan be done using oligo-dT cellulose spin columns, oligo-dT/ magnetic beads and coated plates.The principle involved at isolation of mRNA relies on base pairing between the polyA residuesat the 3’ end of most mRNA, and the oligo (dT) residues coupled to the surface of cellulosespin columns or, magnetic beads or, a pre-coated 96 oligo-dT plate.
Independently of efficiency the three kinds of mRNA isolation are available commercially,facilitating the lab work.
Since the mRNA is available, the cDNA can be produced. To produce the cDNA the reactionshould be done using mRNA template and a mix of, primers, reverse transcriptase, solutionof four dNTPs and buffers. Depending on the experiment, ligo (dT)12-18, random hexanu‐cleotides, or gene-specific antisense oligonucleotides can be used as primers for synthesis offirst-strand cDNA [25].
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607177
The correctly native gene synthesis by this method depend on the fidelity of copying mRNAand also on the stability of DNA thus synthetized. Moreover, since mRNA of a gene does nothave the complete transcript of the gene in vivo (intronic regions are dismissed) the synthesizedgene will be smaller than the gene in vivo, but contain just the coding sequences, what couldbe a great advantage for research [9].
5.4.3. Synthesis by PCR
The gene synthesis by PCR, as described first for W. P. C. Stemmer and coworkers werereported having four steps. First the olygos are synthetized, and then the gene is assembled,amplified and cloned. Since single-stranded ends of complementary DNA fragments are filledin during the gene assembly process, cycling with DNA polymerase results in the formationof increasingly larger DNA fragments until the full-length gene is obtained [26].
The classical method involves the use of oligonucleotides of 40nt long that overlap eachother by 20nt. The oligonucleotides are designed to cover the complete sequence of bothstrands, and the full-length molecule is generated progressively in a single reaction byoverlap extension PCR, followed by amplification in a separate tube by PCR with two outerprimers [27].
Variations of the classical approach were done, such as ligation of phosphorylated overlappingnucleotides, modified form of ligase chain reaction combinations with asymmetrical PCR andthermodynamically balanced inside out.
Nevertheless, most of them are based on phosphorylation of oligos at the 5’ ends’, annealingof overlapping ends, filling the gaps by enzymatic extension at 3’ ends and join nicks withDNA ligase. Then the full length double stranded DNA can be cloned on a plasmid/phagevector and multiplied in E. coli or, amplified by PCR, separated on electrophoresis, purifiedfrom gel and cloned [9].
The most commonly synthesized genes range in size from 600 to 1,200 bp although, muchlonger that genes made by connecting previously assembled fragments of fewer than 1,000 bp.In this size range it is necessary to test several candidate clones confirming the sequence of thecloned synthetic gene by automated sequencing methods [23].
6. Cloning vectors
The molecular cloning brings the possibility to isolate, analyze, synthetize and clone individualgenes or segments of DNA, creating a recombinant DNA. After isolated and purified the DNAtarget sequence must be mounted on an appropriate carrier molecule, the cloning vector.
A cloning vector is a small piece of DNA into which a foreign DNA is inserted for transfer orpropagation in an organism, with the ability to self-replicate. The purpose of a vector is toallow efficient high-level expression of cloned genes or still, the need to increase the numberof copies of a recombinant DNA [28].
78Genetic Engineering
6.1. Need to increase the number of copies of recombinant DNA
Besides having a DNA molecule already recombined, single copies are not sufficient toconstruct a recombinant DNA. The in vitro manipulation like, purification and transfer to atarget cell, of a single copy is not possible. Thereby the recombinant construct should bepropagated to increase the copy number. A convenient way to copy such fragments is to usethe replication machinery of an organism, inserting the donor DNA in a cloning vector [29].The essence of molecular cloning is to use restriction nucleases to cut DNA molecules in astarting DNA population (the target DNA) into pieces of manageable size, then attach themto a replicon (any sequence capable of independent DNA replication) and transfer theresulting hybrid molecules (recombinant DNA) into a suitable host cell which is then allowedto proliferate by cell division. Because the replicon can replicate inside the cell (often to highcopy numbers) so does the attached target DNA, resulting in a form of cell-based DNAamplification [11].
6.2. Cloning vectors
In principle, any molecule of DNA that can replicate itself inside a cell system could work asa cloning vector, but many factors as, small sizes, mobility between cells, easy production anddetection mechanism should be considered [28].
The type of host cells used in a particular application will depend mainly on the purpose ofthe cloning procedure. Host cells exploited are modified bacterial, fungal cells (e.g. Yeast), orstill virus, being the bacterial system (e.g. E. coli) the most used due to their capacity for rapidcell division and for attend the major vectors requirements.
The vector may have an origin of replication that originates from either a natural extrachro‐mosomal replicon or, in some cases, a chromosomal replicon [11]. Besides the structure thevectors should contain a sequence that make possible to select the recombinant cells, like amarker gene and in third place they should contain restriction sites into which the DNA canbe inserted [29].
The types of cloning vectors are plasmids, phages, cosmids, phagemids, artificial chromo‐somes, viral vector and transposons. Each of them will be briefly describe in this section.
6.2.1. Plasmideal vectors
Plasmids are small circular double-stranded DNA molecules, which exist in the cell asextrachromosomal units. In a cell, they have the ability for self-replicating, and copy numbersmaintenance. Due to their capacity of copy numbers they can be classified as: single copyplasmids or multicopy plasmids.
The single copy plasmids are maintained as one plasmid DNA per cell, instead the multicopyplasmids that are maintained as 10-20 copies per cell. Another kind of plasmids consists inones that are under relaxed replication control, allowing their accumulation in numbers up to1000 copies per cell, being the used ones as cloning vectors [15].
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607179
The plasmids vectors are designed to work in bacteria cells. An important property in thesevectors is the detection of the same in the host cells. Usually, the detection mechanisms aredone through antibiotic resistance. The host cell strain chosen is sensitive to a particularantibiotic and the plasmid is designed to contain a gene conferring resistance to this antibiotic.Another approach for detection is through β-galactosidase gene complementation in whichthe host cells are mutants containing a β-galactosidase gene fragment and plasmid vector aredesigned to contain a different fragment of the same gene. By this way, after transformationfunctional complementation occurs and the host cells, which incorporate the plasmid arecapable of β-galactosidase production.
The functional β-galactosidase activity can be accessed by conversion of a colorless substrate,Xgal (5-bromo, 4-chloro, 3-indolyl β –D- galactopyranoside) to a blue product [11]. The bothmethods are efficient for clone’s selection, and their use depends on individual’s preferences.According to P. K. Gupta (2009) [15], there were three phases of plasmid development cloningvectors. The first included the plasmids pSC101, ColE1 and pCR1, which are naturallyoccurring plasmids, and not suitable for efficient cloning, since plasmid can transfer the genethrough bacterial conjugation or can be integrated in the bacterial genome having no accessibledetection system. Other disadvantage lies on having no more than two restriction sites forcloning.
The drawbacks of naturally occurring plasmids were overlapped by pBR313 and pBR322.pBR313 was too large having fifty percent of its sequences being non-essential. The sizereduction brought the pBR322, which was largely used for many years.
The second phase relies on reducing the plasmids sizes, because the transformation efficiencyand vector size have a proportional inverse relation. Thus, variations of the pBR322 appeared,including pAT153, pXf3, pBR327, etc. This plasmid vectors incorporate the selection mecha‐nism of antibiotic resistance (described above).
The third phase involves incorporation of sequences for alpha-complementation selection(described above); incorporation of sequences from single strand M13 phage, for sequencingtemplates production; and, also integration of promoters’ sequences, for in vitro transcriptionor expression of large amounts of foreign proteins. In this phase, plasmids like pUC, pGEM,M13, were developed.
Nowadays, there are a lot of plasmids commercially available that can be purchased dependingon the application needs.
6.2.2. Lambda phage vectors
A bacteriophage lambda is a bacterial virus that infects E. coli. Its utility as a cloning vectordepends on the fact that not all of the lambda genome is essential for its function [1]. Thelambda genome has the left-hand region with essential genes for the structural proteins andthe right-hand region has genes for replication and lysis, while the middle region has the genesfor integration and recombination, which are non-essentials.
80Genetic Engineering
There are two possible types of lambda vectors: the insertion vector and the replacementvector.
The insertion vector has only a single recognition site for one or more restriction enzymes,enabling the DNA fragment to be inserted into the lambda genome. The lambda particleintegrates DNA molecules between 37 and 52kb, and to adapt longer inserts is necessary toremove some of lambda genome. The region for replacement is the middle one where, more23 kb of foreign DNA can be inserted. This vector is known as replacement vector [28].
The replacement vector cannot be integrated into the host cells chromosome being necessaryto use a helper phage to provide integration and recombination functions. On the other hand,this vector has two restriction sites, having a whole section of phage genome being replacedduring cloning [1].
6.2.3. M13 phage
M13 is filamentous bacteriophages that infect specific E. coli. Your attractive as a cloning vectorconsists in its genomes contain the desirable size for a potential vector (less than 10kb); doesnot kill the host when progeny virus particles are released and thus, is easily prepared froman infected E. coli cells culture. Besides, M13 is used as cloning vector to make single strandedDNA for sequencing and mutagenesis approaches.
The M13 genome is a single-stranded DNA molecule with 6407bp in length. This bacterio‐phage only infects bacteria carrying the F-pili (fragile protein appendages found on conjuga‐tion-proficient cells), being male-specific. When the DNA enters the cell, it is converted to adouble-stranded molecule known as replicative form, which is a template for making about100 copies of the genome. At this point replication becomes asymmetric, and single-strand‐ed copies of the genome are produced and extruded as M13 particles. The property of donot lyse the host cell brings a DNA resource, although growth and division are slower thanin non-infected cells [1, 11, 28].
6.2.4. Cosmids
Cosmids are plasmid particles into which certain specific DNA sequences, namely those forcos sites, are inserted. The goal of these vectors development is to cloning of large DNAfragments (up to 47kb in length). They are made up of plasmid sequences joined with lambdavectors sequences, trying to conjugate the properties of this both vectors in one (beingtransfected as a lambda vector by packaging/ infection mechanism and behaving as a plasmidwhen introduced into an E. coli cell).
The advantages consist of a highly efficient method of introducing the recombinant DNA and,a cloning capacity twofold greater than the best lambda replacement vectors. On the otherhand, the gains of using cosmids instead of phage vectors are offset by losses in terms of easeto use and further processing of cloned sequences [1].
The methodology to use the cosmid cloning vectors consists in put together the cleaved vectorand the target DNA for cloning, producing concatameric molecules. The concatameric
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607181
molecules are usually generated by first linearizing the cosmid so that each end has cos site.Then the linear cosmid is cut with a BamHI, which generates sticky ends with the overhangsequence GATC. The foreign DNA is also digested with Mbol, which also generates a GATCoverhang. Partial digestion leaves some site uncut and allows large segments of a genome tobe isolated. These segments are mixed with the two halves of cosmid and joined using ligase.Thus, these molecules are packaged into phage heads by mixing with a packaging extract,becoming infectious. E. coli cells are infected with the cosmids, and after infection the cosmidcircularizes and multiply as a plasmid vector [15, 28].
6.2.5. Phagemids
Phagemids combine desirable features of both plasmids and bacteriophages. The constructconsists of a plasmid with a segment of a filamentous bacteriophage, such as M13, having twodifferent origins of replication: the plasmid and the phage origin. The selected phage sequencescontain all the cis-acting elements required for DNA replication and assembly into phageparticles [11, 30].
These vectors allow successful cloning of inserts several kilobases. After E. coli suitable straintransformation with a recombinant phagemid, the bacterial cells are superinfected with afilamentous helper phage, activating the phage origin and the phagemid. The plasmid DNAcreates single stranded DNA, which is secreted into phage particles. These particles contain amix of recombinant phagemids and helper phage. The selection is usually done by β-galacto‐sidase gene complementation and by antibiotic resistance.
Vector pairs that have the phage origin in opposite directions are available, and as a resultsingle stranded DNA representing of both DNA strands are produced. This mixed singlestrand DNA population can be used directly for DNA sequencing, if the primer for initiatingDNA synthesis is designed to bind specifically to sequences of phagemid adjacent to thecloning site [11, 30].
Both cosmids and phagemids are characterized as hybrid vectors.
6.2.6. Chromosome Bacterial Artificial (BAC)
A bacterial artificial chromosome (BAC) is a single copy bacterial vector based on a functionalfertility plasmid (F-plasmid) of E. coli, which can accept very long inserts of DNA between300-350kb and allows the maintenance of many structural characteristics of the native genome.BAC vectors are superior to other bacterial system, due to the F factor, which has genesregulating its own replication and controlling its copy number. These regulatory genes are oriSand repE, mediating unidirectional replication and parA and parB, maintaining the copynumber to one or two per cell. The cloning segment includes the lambda bacteriophage cosNand the P1 loxP sites; two cloning sites (HindIII and BamHI); and, several C+G rich restrictionenzyme sites (Not I, Eag I, Xma I, Sma I, Bgl I and Sfi I) for potencial excision of the inserts. Thecloning site is flanked by T7 and SP6 promoters for generating RNA probes for chromosomewalking and for DNA sequencing of the inserted segment at the vector-insert junction. The
82Genetic Engineering
CosN and loxP sites provides convenient generation of ends that can be used for restriction-site mapping to arrange the clones in an ordered way [31].
Besides the maintenance of large DNA inserts, BAC has structural stability in the host, highcloning efficiency and easy manipulation of cloned DNA, being largely utilized for construc‐tion of DNA libraries from complex genomes and subsequent rapid analysis of complexgenome structure [31].
For recombination with DNA inserts, after enzymatic digestion DNA ligase are used. Trans‐formed suitable E. coli was carried out by electroporation, and the competent cells are culti‐vated first with gentle shaking on liquid medium and then spreading to LB plates. The selectionof recombined cells is done by hybridization procedures.
6.2.7. Animal virus
Viral vectors are commonly used to deliver genetic material into cells for gene therapy dueto specialized molecular mechanisms to efficiently transport their genomes inside the cellsthey infect. This process can be performed inside a living organism (in vivo) or in cell culture(in vitro), being frequently used to increase the frequency of cells expressing the trans‐duced gene [32].
The first use of vector virus for cloning was based on simian virus 40 (SV40), a polyomavirusoriginated of rhesus macaque, being a potent DNA tumor virus infecting many types ofmammal cells in culture. The SV40 genome is 5.2 kb in size and contains genes coding forproteins involved in viral DNA replication, and genes coding for viral capsid proteins. Due topacking limitations, cloning with SV40 involves replacing the existing genes with the foreignDNA [32-34].
The other kinds of virus used for mammals’ gene clones are adenoviruses, papillomaviruses,adeno-associated virus, herpes simplex virus (HSV), poxvirus and more recently retroviruses.Adenoviruses came to solve the size of insert drawback of SV40, enabling the cloning of DNAfragments up to 8kb. On the other hand, due to its larger genome, adenoviruses are difficultto handle. Expression can be transient and the in vivo transfection can be impaired due toimmune response.
Papillomaviruses also have a high capacity for inserted DNA with the advantage of stabletransformed cell line.
Adeno-associated virus has this name because it is often found in cells that are simultaneouslyinfected with adenovirus. To complete the replication cycle the adeno-associated virus usesproteins already synthesized by adenovirus, which acts like a helper virus. Lack of helper virusmade the genome of adeno-associated virus integrate to host DNA. The major advantage ofthis vector consist of a defined the insertion site, always in the same position, being importantin researches that cloning gene needs rigorously check such as gene therapy.
The herpesviruses include infections human viruses as herpes simplex virus (HSV), most usedlike a vector. The HSV is an enveloped double-stranded DNA, with 152kb, having advantages
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607183
like larger foreign DNA carrying; high transduction efficiency and, potential to establishlatency.
Poxvirus vectors are double-strand DNA with 200kb in the core and carrying up to 25kb oforeign DNA. Gene is stably integrated into the virus genome resulting in efficient replicationand expression of biologically active molecules.
Many viruses kill their host cells by infection, so special artifices are needed if anything otherthan short-term transformation experiments is desirable. Bovine papillomavirus (BPV), whichcauses warts on cattle, is particularly attractive because they have an unusual infection cyclein mouse cells taking the form of a multicopy plasmid with about 100 molecules present percell. This infection does not bring the death of cell and, BPV molecules are passed to daughtercells during mitosis.
The most used viral vectors are the retroviruses, infectious viruses that can integrate intotransduced cells with high frequency, inserting the foreign DNA at random positions but, withgreat stability. They can be replicated-competent or replication-defective.
Replication-competent viral vectors contain all necessary genes for virion synthesis, andcontinue to propagate themselves once infection occurs. These vectors can integrate an insertedabout 8–10 kb, limiting the introduction of many genomic sequences. This made replication-defective vectors the usual choice. These vectors had the coding regions replaced with othergenes, or deleted. These viruses are capable of infecting their target cells but they fail tocontinue the typical lytic pathway that leads to cell lysis and death.
The viral genome in the form of RNA is reverse-transcribed when the virus enters the cell toproduce DNA, which is then inserted into the genome at a random position by the viralintegrase enzyme. The vector, now called provirus, remains in the genome and is passed onto the progeny of the cell when it divides. The site of integration is unpredictable, which canpose a problem; therefore, the principal drawback of retrovirus vectors involves the require‐ment for cells to be actively dividing for transduction, being widely used in stem cells. Greatexamples to overcome this disadvantage are lentiviruses vectors.
The lentivirus is a subset of retrovirus with the ability to integrate into host chromosomes andto infect non-dividing cells. Lentivirus vector systems can include viruses of non-human origin(feline immunodeficiency virus, equine infectious anemia virus) as well as human viruses(HIV). And for safety reasons lentiviral vectors never carry the genes required for theirreplication, preventing the occurrence of a wildtype-potentially infectious virus [32-34].
6.2.8. Transposons
DNA transposons elements are natural genetic elements residing in the genome as repetitivesequences that move through a direct cut-and-paste mechanism. This process is independentof previously recognized mechanisms for the integration of DNA molecules and occurswithout need of DNA sequence homology. Thus, they can be used as tools from transgenesisto functional genomics and gene therapy.
84Genetic Engineering
Transposons are organized by terminal inverted repeats (ITRs) embracing a gene encodingtransposase necessary for relocation. Transposons move through a “cut-and-paste” mecha‐nism, known as transposition, which involves excision from the DNA and subsequentintegration into a new sequence environment [35, 36].
The development of transposable vectors is based on a plasmid system, with a helper plasmid(expressing the transposase) and a donor plasmid (with terminal repeat sequences embracingthe foreign gene) [36].
6.3. Importance of promoters
The promoters are defined as cis-regulatory elements responsible for the control of transcrip‐tional machinery and determination of its level and specificity, marking the point at whichtranscription of the gene should start, and regulating the transcription. Promoters containproximal elements, involved in the formation of the transcription complex; and, majorelements that give cell specificity of protein expression [37, 38].
For long term transgenic expression in vivo or tissue specific expression, the transcription ofthe foreign gene should be controlled for promoters, which in this case are inserted on cloningvectors [37].
Approaches requiring a high ubiquitous expression of the transgene can be accomplished withnon-tissue specific promoters. These promoters are actives in almost all of cell types, ensuringthe foreign gene expression in all organism tissues. Examples of these promoters are metallo‐thionein gene promoter, EF1 gene promoter, CMV early gene promoter, human H2K genepromoter, 3-methylglutaryl CoA reductase gene promoters, and others.
On the other hand, to restrict transgene expression to the target tissue the promoters used aretissue-specific. These promoters can direct the transgene expression to lung, epithelia, liver(albumin gene promoter), pancreas (amylase promoter), muscles (truncated muscle creatinekinase - MCK), neural cells (synapsin 1), mammary gland and cardiac cells (troponin Tpromoter), and so on [38]. Promoters used in cloning vectors should be sufficiently short to becloned in a gene transfer vector.
Besides the use of tissue-specific promoters, another kinds of promoters are the inducible ones,which transcription can be selectively activated. These promoters respond to specific tran‐scriptional activators are: transcriptional activators regulated by small molecules; intracellularsteroid hormone receptors; and, synthetic transcription factors in which dimerization iscontrolled by antibiotics.
The promoters for transcriptional activators regulated by small molecules are based on the useof transcription factors that change their conformation upon binding one small chemicalmolecule (e.g. Tet repressor – TetR). The promoters for intracellular steroid hormone receptorsact when hormone analogs are ligated to the hormone’s modified receptors. Synthetictranscription factors in which dimerization are ones that in the presence of antibiotics tethersthe transcriptional activation [37, 38].
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607185
7. Practical application of genetic Engineering and cloning: From
transgenic animal models until cloning animal
Transgenic animal technology and the ability to introduce functional genes into animals arepowerful and dynamic tools of genetic engineering. The genetic engineering field allows stableintroduction of exogenous genetic information into any live organism, enhancing existing or,introducing entirely novel characteristics. The cloning technology is closely related withtransgenic, being used as a tool for genetic engineering of an animal.
Together these technologies can be used to dissect complex biological process, like in vivo studyof gene function during development, organogenesis, aging, gene therapy, and epigeneticsstudies. Besides, there are a lot of commercial applications like, model for human diseases,pharmaceutical biotechnologies development, and reproduction of a valuable animal.
7.1. A sheep named Dolly: Cloning
In 1997, Wilmut and coworkers announced Dolly production, which was the first mammalcloned from adult cells. In this experiment Dolly was born after reconstruction of 277 embryoswith mammary gland cells.
Her birth at 5 July 1996 in Scotland brought huge excitement of the scientific world, beginninga biological revolution. The fact of Dolly has been created from adult differentiated cellsshowed the possibility not imagined before: dedifferentiation of already committed somaticcells, which brings a lot of repercussion. After Dolly, the differentiated cells cloning wasachieved in a lot of species like, bovines, murines, caprines, swines, felines and canines [39-46]
7.2. What is cloning?
The definition of clone consists in the reproduction of genetically identical organisms,naturally or artificially, by asexual reproduction (without spermatozoa). The word “clone”comes from the greek word “klon”, that means twig. With these characteristics clone for someorganisms is a physiological asexual way of reproduction (e.g. bacteria and yeast). Thisconception, after Dolly’s production went further, becoming the production of geneticallyidentical live organism through nuclear transfer techniques. This defines clone to a process inwhich cellular material from a DNA donor is transferred to an egg whose own DNA has beenremoved, resulting after some procedures in embryo genetically identical to DNA of originalcell clone.
The origins of nuclear transfer remount discoveries with amphibians by Spemann (1938), whodemonstrated that nuclei of newt salamanders are pluripotent up to eight-cell stage, leadingintensive studies with nuclear transfer in Rana pipiens and Xenopus laevis, attempting tounderstand the nuclei participation of differentiated cells in reprogramming. Studies by Bringsand King (1952) showed that amphibian oocytes receiving blastula nuclei could be reared tomaturity [47].
86Genetic Engineering
During 60 and 70 decades, nuclear transfer was done mostly in amphibian, leading to clonesproduction from intestinal larvae cells, being the first evidence that differentiated cells keepthe potential to form all tissues of an organism. [48].
In mammals the first nuclear transfer studies were done in mice, in which Illmensee and Hoppe(1981) reported that this technique could be used to produce mice clones from embryo cells.In domestic animals, Willadsen (1986) published the first report with lamb clones production.This accomplish was confirmed after with bovines, rabbits, swine, and others. And, at 1996Dolly brought the accomplishment of mammal nuclear transfer form adult cells [48].
Nowadays, a lot of cloned animals could be produced, which besides the commercial interestof reproducing some valuable animal, made the technique used for research like reprogram‐ming mechanism and epigenetics controls.
7.3. Producing a clone: Technical
Technically to produce a clone from nuclear transfer the majority of protocols are based onthese steps: preparation of cytoplasm receptor; oocyte enucleation; preparation of nuclei donorcells; embryo reconstruction; artificial activation; embryos culture; and, embryo transfer. Eachof them will be briefly described below.
7.3.1. Methodology, advantages and disadvantages
Initially to produce a clone animal by nuclear transfer is necessary to prepare the receptorcytoplasm. The receptor cytoplasm is a cell which nuclei was removed by in a process knownas enucleation, being the most used cell the female egg: the oocyte. The oocyte can be used ina lot of division estate being, actually used in metaphase II.
To obtain the oocyte at metaphase II, first, or they are aspirated from ovaries from slaughter‐houses or by Ovum Pick Up, ultrasound guided (used for domestic animals), being obtainedbefore metaphase II, needing in vitro maturation or, being collected already at this estate, afterin vivo maturation (mostly used for laboratory animals).
When maturation is needed, the oocytes are recovered from antral follicles at prophase I estateor germinate vesicle, and are maturated in temperature, medium and time specific, insideincubators, having the control of CO2 tension. The two kinds of procedure to obtain a meta‐phase II oocyte, have its advantages and disadvantages. The in vivo maturation, are capable ofbetter quality oocyte production, but depending on the species, it cannot be achieved, needingthe in vitro maturation. The in vitro maturation besides not be the natural way of reproduction,brings good and quality results too, being largely used.
The second step consists of oocyte enucleation. Besides a lot of methodologies have beendeveloped, the most used way to remove the nuclei from the oocyte is micromanipulationprocedures.
Before, to prepare the oocytes for enucleation, the cumulus cells from the maturated ones areremoved, which can be done mechanically by a tube agitator (vortex); with pipettes sized asthin as the oocyte; or, chemically by hialuronidase. After, the oocytes are carefully selected,
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607187
based on the presence of the first polar body (checking if the metaphase II was achieved), andcytoplasm morphology.
The micromanipulation procedure for enucleation involves first fixing the mature oocyte bya holding pipete. To remove the nuclei, the first polar body is used to reference of the meta‐physary plate (nuclei). At metaphase II the chromatin remains at the oocyte periphery, closeto the polar body. The enucleation is done by an enucleation pipete with a bevel-shaped tip,which penetrates the pellucid zone (PZ) aspirating the first polar body e part of the cytoplasmattached to this structure.
Another enucleation method is the oocyte bisection [49]. In this case the PZ is removed andthe oocytes are sectioned by a micro-blade in two halves, being removed 50% of the cytoplasm.The half with the nuclei is discarded and the other one used for nuclear transfer.
To check the efficiency of enucleation, the enucleated oocytes can be stained with DNAfluorescent dyes using in most of the cases Hoechst 33342 (H342), which need exposure ofultra-violet (UV) light to be verified.
This procedure depending on the time of exposure can compromise the oocytes viability.Trying to minimize this effect the exposure only of the removed cytoplasm and polar body toUV light can be done. The presence of chromatin in this material indicates the success ofenucleation. There are protocols not involving fluorochromes like the use of demecolcin,incubating the oocytes 1-2 hours, in a medium containing this substance. This procedurecreates a protrusion at oocyte membrane where the chromatin is localized [48].
The amount of cytoplasm removed at enucleation process direct interferes in embryosdevelopment rates. The less amount of cytoplasm removed, better rates of embryo develop‐ment are achieved. Usually, the enucleation for bovines has an efficiency rate between 50-70%[46].
The third step is preparation of nuclei donor cells, which depends of the cell type and thetechnique used for nuclei transfer. The donor cells can be originated from embryonic, fetal oradult cells. When using embryonic cells, the PZ of embryos are removed by enzymaticdigestion, acid solution or mechanically. The embryonic mass should be held on a calcium andmagnesium free solution, facilitating the blastomeres disintegration. If the donor cell was fetalor adult mostly fibroblasts are used due to easy culture. A primary culture is done by a biopsyfrom a skin fragment. The cells are held on culture until the third passage, at least, due tohomogeneity and specific cells reaching on the culture, more than the third passage cells canbe used as nuclei donor too.
Besides a lot of experiments have been realized to determine which somatic cell type wouldbe the most appropriated for cloning, until now is not yet known if some kind of cell are mostadvantageous for nuclear transfer [48].
The fourth step is embryo reconstruction consisting of place the nuclei from the donor cellinside the enucleated oocyte. This can be achieved by microinjection or membranes fusion,whereas the first has low results [46]. Using the fusion method with micromanipulators help,each cell is introduced at the perivitelline space of the enucleated oocyte. Then the fusion can
88Genetic Engineering
be done by electric pulses (electrofusion), liposomes, polyethylene glycol, or still, by inacti‐vated viruses. The electrofusion are the mostly used. In this method, the complexes receptor-donor nuclei are positioned at electrofusion chamber, where they are submitted to two electricpulses with low conductance, preventing heat dispersion. These pulses induce the membranefusion incorporating the cell donor nuclei at the receptor cytoplasm.
The fifth step consists of artificial activation, which involves degradation of enzymaticcomplexes responsible for oocytes kept at metaphase II, being needed for accomplish themeiotic process initiating the embryonic development. At physiologic conditions, this isachieved with spermatozoa. But at cloning process, without the spermatic cell, chemically orphysical methods are used (ethanol, electric pulses, calcium ionophore and, strontiumchloride).
The activation moment of oocyte in relation with the nuclear transfer moment have importantconsequences at the chromatin integration and remodeling; viability and, embryo develop‐ment [48].
The sixth step consists of embryo culture, in which the reconstructed and activated embryosare cultivated at CO2 incubators, until the blastocyst stage (species time dependent). Theculture conditions are similar with in vitro fertilization conditions, whereas cloned embryosare more sensitive to cryopreservation, and do not pass through expansion phase whenblastocyst stage are achieved. The PZ rupture, done by enucleation process, made the expan‐sion estate coincide with hatch estate, and at this estate the embryos are transferred tosynchronized female receptors, and after gestation and parturition or caesarean, clonedanimals are produced [46].
7.4. What is a transgenic animal?
A transgenic animal consists of an animal whose genetic material has been altered usinggenetic engineering techniques. Foreign DNA is introduced into the animal, using recombi‐nant DNA technology, and then must be transmitted through the germ line so that every cell,including germ cells, of the animal contains the same modified genetic material [32, 50].
S. N. Cohen and H. Boyer generated a functional organism that combined and replicatedgenetic information from different species, creating the first genetic modified organism in 1973.In 1974 R. Jaenisch created the first genetically modified animal by inserting a DNA virus intoa mouse embryo showing inserted genes was present in every cell. However the mice did nottransmit the transgene. In 1981 F. Ruddle, F. Constantini and E. Lacy injected purified DNAinto a single-cell mouse embryo and showed transmission to subsequent generations. Duringthe early eighties the technology used to generate genetically modified mice was improvedinto a tractable and reproducible method [51, 52].
7.5. Producing a transgenic: Technical
Currently, the three most widely used procedures for creating transgenic animals are micro‐injection of the cloned gene(s) into the pronucleus of a fertilized egg, injection of recombinant
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607189
embryonic stem cells into embryos, and the use of retroviruses. There are other methods likesperm cells mediated gene transfer; in vivo gene transfer and ICSI-transgenes. These methodswill be briefly discussed below.
7.5.1. Methodology, advantages and disadvantages
The microinjection of foreign DNA directly into the pronuclei of fertilized zygotes is the mostextensively and successfully used method of gene transfer in the mouse. This method was thefirst non-viral method for transgenic animal production. The DNA microinjection to pronu‐cleus has low technical progress, but was disseminated for other species (rabbit, swine andgoats) [53, 54].
To produce a transgenic animal a lot of zygotes are needed which is achieved by femalesuperovulation before mating. For mouse, rats and rabbits the one cell embryos are transpar‐ent, being opaque in swine, goats, sheeps, and cow, due to lipid presence. In case of opaqueembryos they should be centrifuge before the microinjection for concentration of lipids at oneembryo side, allowing the pronuclei visualization.
The disadvantages of this technique are due to exogenous DNA introduced at the pronucleusis strongly mitogenic, leading a lot of embryos microinjected to death. Another disadvantageconsists of integration of foreign DNA in a random manner, being not possible to predict theintegration site and control the number of copies of transgenic integrated DNA. The transgenicproduction by microinjection to pronucleus are 2% for mouse, 0,1-0,5% for pigs, 0,01-0,1% forsheeps and goats and lower for cows [54].
Another used methodology is infection by retroviruses, in which the transgenes can beintroduced by viral infection of preimplantation embryos. Retroviruses have the natural abilityto infect cells and integrate its genome to infected cells. The retroviruses are modified, in whichsome of its genes are substituted by a target gene. Then these reconstructed viruses aretransferred for cells, that after infected synthesize viral protein, secreting viral particles whichcan infect embryonic cells, or primordial germ cells, originating transgenic animals.
The disadvantages came from biosecurity, since this technique works with recombinedviruses. This determines biosecurity rules to be carefully followed, preventing this vectordissemination. One advantage is the use of this method is for gene therapy, transferring themodified viruses for somatic cells of the patients. And another advantage came with lentivi‐ruses uses that are successfully used for gene transfer (see item 5 for advantages and disadvantagesof this virus).
The gene transfer by injection of recombinant embryonic stem cells into embryos is one of themost useful when is necessary to select for rare integration events or when is necessary anchimeric animal production [53].
Embryonic stem cells (EST) are cell lineages obtained from initial embryos estate, like morulaor blastocyst. These cells have pluripotency capacity, being capable of participation activelyof all organism tissue production, including the gametes. The technique for gene transferconsists in transfect EST with exogenous DNA; and the ones transfected after been selected,
90Genetic Engineering
are introduced into embryos. The resulting animals are chimeric and mosaics for the transgene,and if the germ cells have integrated the transgene, this can be passed for the progeny.
The disadvantages of this methodology are the impossibility of chimeric animals from otherspecies besides mouse; transmit the transgene to their progeny. Being an advantage when thetransgene requires homolog recombination.
Another ways of transgenic animals production are being used like, the use of gametes cellsto transgenic animal production are already achieved. The oocytes uses do not generatedenough transgenic animals, being the spermatozoon most appropriated cells for transgenesis[54].
The Sperm-mediated gene transfer (SMGT) enables the production of transgenic animals byexploiting the ability of sperm cells to bind and internalize exogenous DNA. The SMGT hasbeing an easy and low cost method for transgenic animals production, in which the simpleincubation of the exogenous DNA with the sperm, follow by artificial insemination or IVFprocedures, can result in an transgenic animal. Although, a lot of transgenic animals producedby this technique, there are high number of studies with low reproducibility and the reasonfor this are unknown.
To pass through this low reproducibility the spermatic cell are been used for integration of thenew genetic material by intracytoplasmic sperm injection (ICSI-transgenesis), method largelyand successfully used for mammalian species other than mouse [53].
The in vivo transfer of a transgene can be done by the injection of the exogenous DNA to testiclesor still blood veins, bringing good results. The revolutionary of this method came withproduction of in vitro sperm stem cell, from many species. After cultivated this primordialsperm cells are transfected with the exogenous DNA, then the recipient animal are treated fordecrease its physiological sperm production, and the transfected cells are injected troughtesticle efferent duct. Thus, after the spermatogenesis this male can be used for transmissionof transgenic sperms.
7.6. Transgenic animals such as experimental models
Genetically modified animals currently being developed can be different broad intendedpurpose of the genetic modification: to improve animal production; xenotransplantation; toproduce proteins intended for human therapeutic use; to improve animals' interactions withhumans trough hypo-allergenic pets; to improve animal health by disease resistance animals;and, to research human diseases with the development of animal models [50].
The animal models production are used in pathologies or syndromes caused by inactivationor dysfunction of a determinate gene, being possible to delineate strategies and prepare theequivalent genetic modification in a homologous gene of the animal [55].
The use of animal models to research human disease, are done because of similarity to humansgenetics, anatomy, and physiology or for being easier to have a lot of conditions developed inmany transgenic animals, manipulating just one variable each time, which corroborate withstatistical analyses of the results.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607191
Extensive research for human diseases have been done with rats, mice, gerbils, guinea pigs,and hamsters, being the mice the mostly used due to genomic similarities to human and easyand developed handle and production methods; low cost; and, high reproductive rates.
Although these advantages sometimes the small size of mice leads to challenges in the designand application of instrumentation for physiological measurements, being possible to intro‐duce transgenes into larger mammals and also fish like genetically modified zebrafish. Theadvantages using model experimental organisms larger than mice are easier physiologicalassessments and besides, provide alternatives when manipulation of the mouse genome doesnot produce the phenotype one wishes to investigate. Examples of this situation are providedby the hypertensive response of rats but not mice to forced expression of the REN-2 gene and themore severe spondyloarthropathy produced by B27 and b2-microglobulin transgenes in rats [56].Multiple models of diverse pathologies have been generated, like diabetes, obesity, allergy,cancer, cardiovascular disorders, hypertension, embryo development abnormalities and,reproductive system abnormalities [57].
Sometimes, complex animal models are needed, using coexistent modification of genes troughtechniques like knockout, knock in and knock down. Another approach is the use of newsgenetic modification variants, like inducible expression of transgenes and restriction oftransgene expression at some organs. These techniques can generate animal models that betterfit some human pathology [57].
7.7. How to join cloning with genetic modification: Complementary biotechnologies
The cloning technology together with genetic modification of organism originates a newmethod for animal transgenesis production. Between different areas of application of nucleartransfer, the transgenesis has the major benefits with its advances. Besides laborious, thistechnique allowed more efficiency transgenic production than pronuclear microinjection forruminant animals.
The advantages of this methodology for animal transgenesis production are introduce,functionally delete or subtly genes of interest; produce embryos expressing the transgeneconstitutively not mosaic or chimeric; or still, achieve genetic modifications directed. This lastapproach can be done by insertion of genes in a determined chromosome position.
The genes inactivation by the cloning technique for transgenic production, are achieved bysubstitution from homolog recombination, which largely obtained just in somatic cells.Examples of this approach in pigs was the substitution of β-galactosiltransferase, making itskidneys being resistance to hyperacute rejection in to experimental transplant in non-humanprimates [56].
Another importance of this approach consists of the applications of transgenic farm animals.The major opportunity of this use is biopharming, mostly produced at the milk of transgenicfemale animals, in which cells modified containing important human genes under a promoterfor mammary gland control (for more information of promoters see topic 5). After milk secretionthis therapeutic protein are purified and used for clinical trials to evaluate their safety and
92Genetic Engineering
effectiveness treating human diseases and disorders before gaining regulatory approval. Othertissues examples, used for antibodies production are eggs of chicken or blood of transgeniccattle [58].
8. Future Prospects — Genetic engineering and cloning: A dream or anightmare?
8.1. Emphasis on cloning technology
A clone is an identical copy of the parental material, which is development. Clones from asame cell type will have the same genomic properties, but in multicellular organisms differentcell behavior and phenotypic are observed by the influence of environment. Cloning is not initself a genetic engineering technique as transgenic but both techniques are strongly correlated.Cloning naturally may occurs after a single insemination and if a specific gene is not insertedinto the host genome the resulting animal cannot be considered a genetically modifiedorganism, transgenic [59]
It is well established in literature that clone embryos have a lower total number of cells whencompared to embryos not cloned. Cloned bovine embryos have approximately 9% less cell, andthis rate is 19, 43 and 55% charge respectively for pigs, rabbits and mice cloned embryos and thisrate being positively correlated with the difficulty of performing cloning [60]. The discovery ofcell types that offer better rates cloning is essential to the development and better understand‐ing of the epigenetic molecular mechanisms, nuclear programming and reprogramming; thus,developing more secure and efficient cloning techniques [61]. Use races which have highestcloning success rates as a model, Ex. Japanese Black cattle, could help to find the key points tosolve currently problems as high mortality rate of cloned offspring. In 2003, a study comparedthe size of spermatozoon chromosome telomeres obtained from cloned and not cloned ani‐mals. For both groups was observed that telomere length was maintained throughout animalsage, fact which indicated that cloned animals could be used as breeders [62].
After Dolly′s birth, one glimpses in human medicine to use cloning technologies in genetherapy for tissues and organ replacement without risk of transplant rejection, since the donorwould be the patient himself. For some people, the ability to clone differentiated cell isregarded as the long awaited immortality or as an insult to religious principles. Nowadayscloning assumes a prominent role in the world media among the most controversial issues.However, if would possible to produce healthy cloned offspring with low mortality andwithout genotypic and phenotypic changes, why would clone be so controversial? It isimportant remember that similar process naturally occurs, once the medical literature showsthat one in 250 births produces the identical twins, the current human clone.
Currently cloning technology is in emphasis on science and media, and over the last few yearshas made important advances. However, more knowledge is needed to this technique able toexercise your full potential as a biotechnology in combination with gene therapy and engi‐neering gene.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607193
8.2. Ethical aspects of genetic engineering: Risks and benefits
The generation of the genetic engineering and his major advances in biotechnology as:sequence of complete genome for different plants and animals, creation of transgenic andcloning had the participation of different scientists.
Mendel is considered the father of modern genetics. Their findings were published in 1865 andjust after 50 years (1909) were considered by Wilhelm Johannsen who discovered the genes,which were called before as "elements" by Mendel. In 1953, James Watson and Francis Crickpublished in Nature the discovery of the helical structure of DNA. At 1973, the concept ofgenetic engineering was created by S. Cohen and H. Boyer who published the ability to “cutand paste” the genetic material. However, it was Van Rensselaer Potter the responsible for theterm Bioethics through his book Bioethics: bridge to the future (1971), which was an answerfor the numerous innovations in science. The advances in this field culminated in the an‐nouncement of the complete sequencing of the human genome, being followed by other speciessequencing.
The discoveries and advances of genetic engineering brought an ethical dilemma. For largepopulation, this science has turned into a dangerous knowledge because we are accumulatinginformation faster than ability to manage them. This creates a conflict between ethical princi‐ples and moral norms. Ethics is the part of philosophy that studies the moral ideals andprinciples for human behavior. However, morality is grounded in obedience to customs andhabits. Ethics is different from moral because it is based on moral actions (good and bad)generated by reason. Thus, the concept of bioethics is the applicability of ethics in biologicalscience, its discoveries and advances.
Using these definitions, genetic engineering is not in its bioethical concept harmful to society.Fear of the unknown by lack of information and knowledge has made us to come back atancestors response. The modern society create new myths and rites. However, with techno‐logical advances in genetic engineering the natural boundaries were lost. Now it is the dutyof man to establish and determine these new barriers. Genetic engineering should be ratherlimited and controlled by new ethical and technical barriers.
With industrial development and new technologies we are living in a new era, the biotech‐nology era. With advances, especially in genetics, a new science called genetic engineering thatenables the manipulation (insertion and removal) of genetic material was created. Thistechnique can be applied since one cell organism (bacteria) until complex multicellularorganisms such as farm animals and humans. The genetic engineering, as the industry,searches for new products (life forms) that have greater production efficiency.
The welfare and environmental concerns for genetic engineering were very well discussed byFox, MW (1998)[63]. According to the author, bio processing industry proposes the productionof new forms of energy, synthetic and pharmaceuticals products. However, many promisesof innovation are accompanied by profound discussions regarding the risks and ethicalconcepts. These aspects should not only be discussed by governments and industries, but alsofor the entire population and researchers. Generally genetic manipulation of bacteria andplants, which are transformed into real machines for the production of biological products
94Genetic Engineering
such as hormones, is considered moral and ethical acceptable. This is explained because plantsand bacteria not have the ability to Suffer, to experience pain and emotional distress. Thus,would it be ethically acceptable to use more complex organisms, such as farm animals, asbiological models and bioreactors? Had genetic manipulation any effect on your bodystructure and physiology? If any error occurs during the process, is the birth of animals withanomalies acceptable?
A study conducted in the UK in 1997 by Frewer et al.[64] consider public concepts on appli‐cation of specific and general genetic engineering. The main concepts related to rejection ofgenetic engineering were: personal objections, immoral, unnatural, unethical, harmful,personal worry, negative welfare effects, dangerous risk, tampering with nature and creationof inequalities. The positive aspects were: beneficial, advantageous, necessary and progressive.The data closely relate applicability of genetic engineering with risks and benefits that aredefined by the nature of their application. The activities considered more negative wereassociated with genetic manipulation of animals and humans, confirming results previouslyobtained by previous researchers. The most important for the establishment of a concept is theapplicability of the technology, most humans and animals that plants and microorganisms.The results imply that the public attitudes are defined by the process associated with geneticengineering rather the product of this process. Unnaturalness is one of the most importantconcepts associated with animal and human genetic material. The medical risk is high inbenefice and low in risk and it is considered acceptable. However, non-medical applicationsare low in benefice and high in risk and it is unacceptable.
The complexity of public concepts about genetic engineering generated a hierarchical modelfor the dissemination of information that was proposed in 1990 by Hilgartner [65], which usesconcepts of public debates to enter public opinion. Thus, the ethical and moral concepts ofsociety are fundamental to be associated at genetic engineering to it exerts your completefunction and optimal use. The public ethical concept must be considered particularly to geneticengineering involving animals and humans.
Moreover, models of diffusion of knowledge about risks and benefits of genetic engineer‐ing to society should be applied. Only with more information and more knowledge you canget over rampant enthusiasm and ideological fear. So some paradigms about geneticmanipulation will be clarified. The biggest problem in genetic is not to make changes in theDNA, but to be faithful to a principle which are common to all men, of all cultures andresponsible for perpetuating human and natural environment; therefore more important thanany gene is ethics [66]. So, to finish this chapter there is no better phrase than: ethics isresponsible to regulate and coordinate the genetic engineering, as also occurs in the othersbranchs of science.
Acknowledgements
FAPESP, CNPq and CAPES
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607195
Author details
Mariana Ianello Giassetti*, Fernanda Sevciuc Maria*,Mayra Elena Ortiz D’ávila Assump??o and José Ant?nio Visintin
Laboratories of in vitro fertilization, cloning and Animal Trasngenesis, Department of Ani‐mal Sciences, Veterinary Scholl, University of Sao Paulo – Sao Paulo, Brazil
*The first two authors contributed equally
References
[1]Nicholl DST. An Introduction to Genetic Engineering: Cambridge University Press;2008.
[2]Lewin B. Genes 8: Pearson Prentice Hall; 2004.
[3]Satya P. Genomics and Genetic Engineering: New India Publishing Agency; 2007.
[4]Brown TA. Genomes: Wiley-Liss; 2002.
[5]Shapiro, J. L.; MacHattie, L. E.; Ippen,G. I. K.; Beckwith, J.; Arditti, R.; Reznikoff, W.;Mac-Gillivray, R. The isolation of pure lac operon DNA. Nature, 1969, 224:768-774.
[6]Korman, A. J. Knudsen, P. J; Kaufman, J. F.; Strominger, J. L. cDNA clones for theheavy chain of HLA-DR antigens obtained after immunopurification of polysomesby monoclonal antibody. Proceedings of National Academy of Science of UnitedStates of America.1982, 79:1844-1848.
[7]Maxam, A. M.; Gilbert, W. A new method for sequencing DNA. Proc. Nati. Acad. Sci.USA, 1977, 74 (2): 560-564.
[8]Graham CA, Hill AJM. DNA Sequencing Protocols: Humana Press; 2001.
[9]K GP, Gupta PPK. Biotechnology And Genomics: Rastogi Publications; 2004.
[10]Perkin Elmer. Automated DNA Sequencing Chemistry Guide. Perkin Elmer AppliedBiosystems. 1998, 3-22, 3-27
[11]Strachan T. Human molecular genetics: Taylor & Francis; 2003.
[12]Shinwari J. Automated DNA sequencing. Megabace 1000, 2005.
[13]Kocher TD. PCR, directing sequencing and the comparative approach. PCR methodsand applications. 1992:217.
[14]Rao V. Genome Res. 1994;4:S15-S23.
96Genetic Engineering
[15]Gupta PPK. Molecular Biology and Genetic Engineering: Rajpal And Sons Publish‐ing; 2008.
[16]Pease , A.C.; Solas, D.; Sullivan, E. J.; Cronin, M. T.; Holmes, C. P.; Fodor S.P. A.Light-generated oligonucleotide arrays for rapid DNA sequence analysis. 1994; 91:5022-5026.
[17]Stears RL, Martinsky T, Schena M. Trends in microarray analysis. Nat Med. 2003 Jan;9(1):140-5.
[18]Chaudhuri, J.D. Genes arrayed out for you: the amazing world of microarrays. Medi‐cal Science Monitor, 2005; 11(2):52-62.
[19]Rosa GJdM, da Rocha LB, Furlan LR. Estudos de express?o gênica utilizando-se mi‐croarrays: delineamento, análise, e aplica??es na pesquisa zootécnica. Revista Brasi‐leira de Zootecnia. 2007;36:185-209.
[20]Jurinke C, Oeth P, van den Boom D. MALDI-TOF mass spectrometry: a versatile toolfor high-performance DNA analysis. Mol Biotechnol. 2004 Feb;26(2):147-64.
[21]Gut IG. DNA analysis by MALDI-TOF mass spectrometry. Hum Mutat. 2004 May;23(5):437-41.
[22]Mauger F, Bauer K, Calloway CD, Semhoun J, Nishimoto T, Myers TW, et al. DNAsequencing by MALDI-TOF MS using alkali cleavage of RNA/DNA chimeras. Nucle‐ic Acids Res. 2007;35(8):e62.
[23]Artificial gene syntheis /wiki/Artificial_gene_synthe‐sis#cite_note-Edge81-2 (accessed 1 September of 2012).
[24]Temin HM, Mizutani S. RNA-dependent DNA polymerase in virions of Rous sarco‐ma virus. Nature. 1970 Jun;226(5252):1211-3. PubMed PMID: 4316301. eng.
[25]Sambrook J, Russell DW. Molecular Cloning: A Laboratory Manual: Cold SpringHarbor Laboratory Press; 2001.
[26]Stemmer WP, Crameri A, Ha KD, Brennan TM, Heyneker HL. Single-step assemblyof a gene and entire plasmid from large numbers of oligodeoxyribonucleotides.Gene. 1995 Oct;164(1):49-53.
[27]Young L, Dong Q. Two-step total gene synthesis method. Nucleic Acids Res.2004;32(7):e59.
[28]Clark DP, Pazdernik NJ. Molecular Biology: Academic Press; 2012.
[29]Pratik, S. Genomics and Genetic Engineering New India Publishing, 2007.
[30]Corley RB. A Guide to Methods in the Biomedical Sciences: Springer; 2004.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607197
[31]Shizuya H, Birren B, Kim UJ, Mancino V, Slepak T, Tachiiri Y, et al. Cloning and sta‐ble maintenance of 300-kilobase-pair fragments of human DNA in Escherichia coliusing an F-factor-based vector. Proc Natl Acad Sci U S A. 1992 Sep;89(18):8794-7.
[32]BUY hpucbth. BUY, http://people.ucalgary.ca/~browder/transgenic.html. (accessed 3September of 2012.
[33]Working with viral verctor http://www.stanford.edu/dept/EHS/prod/researchlab/bio/docs/Working_with_Viral_Vectors.pdf (accessed 3 September of2012.
[34]Gene Clonning, chapter 7. /genecloning/pdfs/chapter7.pdf. (accessed 3 September of 2012.
[35]Grabundzija I, Irgang M, Mátés L, Belay E, Matrai J, Gogol-D?ring A, et al. Compara‐tive analysis of transposable element vector systems in human cells. Mol Ther. 2010Jun;18(6):1200-9.
[36]Meir YJ, Wu SC. Transposon-based vector systems for gene therapy clinical trials:challenges and considerations. Chang Gung Med J. 2011 2011 Nov-Dec;34(6):565-79.37.
[37]Giaca, M. Gene Therapy, Springer, 2010.
[38]Pinkert, C.A., Transgenic Animal Technology. A laboratory handbook. 2nd Ed. Aca‐demic press, 2002.
[39]Mello, M.R.B. ; Caetano, H. V. A.; Marques, M. G.; Padilha, M. S.; Garcia, J. F.; As‐sump??o, M.E.O.A.; Lima, A. S.; Nicácio, A. C.; Mendes, C. M.; Oliveira, V. P.; Visin‐tin, J. A. Production of cloned calf from fetal fibroblast cell line. Brazilian Journal ofMedical and Biological Research, 2003, 36: 1485-1489.
[40]Kato, Y.; Tani, Y.; Sotomaru, Y.; Kurokawa, K.; Kato, J.; Doguchi, H.; Yasue, H.; Tsu‐noda, Y. Eight calves cloned from somatic cells of a single adult. Science,1998, 282:2095-2098.
[41]Onishi, A.; Iwamoto, M.; Akita, T.; Mikawa, S.; Takeda, K.; Awata, T.; Hanada, H.;Perry, A.C. Pig cloning by microinjection of fetal fibroblast nuclei. Science, 2000, 289:1188-1190.
[42]Shin, T.; Kraemer, D.; Pryor, J.; Rugila, J.; Howe, L.; Buck, S.; Muphy, K.; Lyons, L.;Westhusin, M. A cat cloned by nuclear transplantation. Nature, 2002, 415: 6874-6859
[43]Wakayama, T.; Perry, A.C.; Zuccotti, M.; Johnson, K.R.; Yanagimachi, R. Full-termdevelopment of mice from enucleated oocytes injected with cumulus cell nuclei. Na‐ture, 1998, 23:369-74.
[44]Woods, G. L.; White, K. L.; Vanderwall, D. K.; Li, G.; Aston, K. I.; Bunch, T. D.; Meer‐do, L. N.; Pate, B. J. A Mule Cloned from Fetal Cells by Nuclear Transfer. Science,2003, 22: 1063.
98Genetic Engineering
[45]Lee, B.C.; Kim, M.K.; Jang, G. O. H. J.; Yuda, F.; Kim, H.J.; Shamim, M.H.; Kim, J.J.;Kang, S.K.; Schatten, G.; Hwang, W.S. Dogs cloned from adult somatic cells. Nature,2005, 436: 641-646.
[46]Mello, M. R. B. Clonagem em Bovinos. In: Palhan, H.B. (ed.) Reprodu??o em Bovinos.Fisiopatologia, terapêutica e Biotecnologia. 2nd Ed. LF livros, 2008. p.225-233
[47]Foote, H.R. Historical Perspective. In: Cibelli, J.; Lanza, P.R.; Campbell, K.H.S.; West,M.D. (ed.) Principles of Cloning. Academic Press, 2002. p. 3-14
[48]Bordignon, V.; Henkes, L.E. Clonagem Animal: a busca da amplifica??o de cópias ge‐neticamente modificadas. In: Collares, T. (ed.) Animais Transgênicos. Princípios emétodos. S?o Carlos, Suprema, 2005. p137-166.
[49]Vajta, G.; Lewis, I.M.; Trounson, A.O.; Purup, S.; Maddox-Hyttel, P.; Schmidt, M.;Pedersen, H.G.; Greve, T.; Callesen, H. Handmade somatic cell cloning in cattle: anal‐ysis of factors contributing to high efficiency in vitro. Biology of Reproduction, 2003,68: 571-578.
[50]Transgenic Animals /wiki/Transgenic_animal#Transgen‐ic_animals (accessed 3 September of 2012).
[51]Genetically modified mouse /wiki/Genetically_modi‐fied_mouse. (accessed 1 September of 2012).
[52]Trangenesis history /transgenesis-history.php. (Ac‐cessed 1 September of 2012).
[53]Nagy, A.; Gertsenstein, M.; Vintersten, K.; Behringer, R. Manipulating the mouse em‐bryo. A laboratory manual, 3rd ed. Cold Spring harbor Laboratory Press, 2003.
[54]Houdebine, L.M. Métodos de gerar animais transgênicos e controle da express?ogênica In: Collares, T. (ed.) Animais Transgênicos. Princípios e métodos. S?o Carlos,Suprema, 2005. p 81-113.
[55]Montoliu, L.; Lavado, A. Animais transgênicos na biologia, na biomedicine e na bio‐tecnologia. In: Collares, T. (ed.) Animais Transgênicos. Princípios e métodos. S?oCarlos, Suprema, 2005. p 114-136.
[56]Williams RS, Wagner PD. Transgenic animals in integrative biology: approaches andinterpretations of outcome. J Appl Physiol. 2000 Mar;88(3):1119-26.
[57]Sutovsky, P. Somatic Cell Nuclear Transfer, Springer, 2007, 591.
[58]Williams, R. S.; Wagner, P. D. Transgenic animals in integrative biology: approachesand interpretations of outcome J. Appl. Physiol, 2000, 88: 1119–1126.
[59]Gordon I. Reproductive Technologies in Farm Animals: CABI; 2005.
[60]Houdebine LM. Cloning by numbers. Nat Biotechnol. 2003;21(12):1451-2.
Genetic Engineering and Cloning: Focus on Animal Biotechnology
/10.5772/5607199
[61]Yanagimachi R. Cloning: experience from the mouse and other animals. Mol Cell En‐docrinol. 2002;187(1-2):241-8. 62.
[62]Miyashita N, Shiga K, Fujita T, Umeki H, Sato W, Suzuki T, et al. Normal telomerelengths of spermatozoa in somatic cell-cloned bulls. Theriogenology. 2003;59(7):1557-65.
[63]FOX MW. Genetic Engineering Biotechnology 1: Animal Welfare and EnvironmentalConcerns. Applied Animal Behaviour Science. 1988;20:83-94. Epub 94.
[64]Frewer LJ, Howard C, Shepherd R. Public concerns in the United Kingdom aboutgeneral and specific applications of genetic engineering: risk, benefit, and ethics. SciTechnol Human Values. 1997;22(1):98-124. PubMed PMID: 11654686. eng.
[65]Hilgartner S. The Dominant View of Popularization: Conceptual Problems, PoliticalUses. Social Studies of Science. 1990;20(3):519-39. Epub 539.
[66]Barth WL. Engenharia Genética e bioética .Rev. Trim., 2005; v.35(149): 361-391.
