实验一 冷原子吸收光谱法测定汞离子
一、 实验目的
1、 巩固原子吸收光谱分析法理论知识。
2、 掌握测汞仪的基本构成及使用方法。
3、 掌握水中汞离子的冷原子吸收测定方法。
二、概述
1、方法原理
仪器根据原子吸收光谱分析的原理即汞子对波长为253.7nrn的共振线上有强烈吸收作用制造的。吸收的大小与汞原子蒸汽的浓度的关系符合比耳定律。
A=lg1/T = lgI0/I = KCL
式中:A一吸光度
I一透射光强度
C一汞蒸汽浓度
T一透光率
I0一入射光强度
K一消光系数
L一吸收光程的长度

由于汞的沸点很低容易挥发,同时汞离子能定量地被亚锡离子还原为金属汞,因而在常温下就可以利用汞蒸汽对253.7nm共振线的强烈吸收来测定溶液中的汞含量。
化学反应式为: Hg2+ + SnCl62- = Hg + SnCl64-
2、仪器
F732—V智能型测汞仪或其它类似仪器。
3、试剂与标准溶液
(l)硝酸:优级纯,分析纯。
(2)盐酸:分析纯。
(3)重铬酸钾:光谱纯。
(4)氯化业锡:分析纯。
(5)汞标准物质:国家一级标准物质。
检定用的汞标准物质要求均匀、稳定、密封在玻璃安培瓶内,有效期为一年,汞浓度值为1.00±0.05mg/ml。由上海测试技术研究所提供。
汞标准物质使用注意事项:
a.使用前注意有效期:从生产日起,一年有效。
b.使用时振摇均匀,保持瓶口清洁,在无汞实验室内方能开启。
C.汞标样放在阴凉干燥处,或冰箱内保存。
d.使用的容器,临用前均需 5%硝酸溶液浸泡 24小时。
(4)汞标准工作溶液:根据工作需要,使用时配制,即对高浓度汞标准溶液,用硝酸重铬酸钾溶液逐级稀释。
(5)硝酸重铬酸钾溶液:称取0.05g重铬酸钾,溶于无汞去离子水,加入5ml优级纯硝酸,再用去离子水稀释到 100ml。
(6)5%硝酸溶液:量取50ml分析纯硝酸,用去离子水稀释至1000ml,供洗涤用。
(7)临用前配制10%氯化亚锡溶液(w/v):称取10g氯化亚锡于小烧杯内,加人20ml浓盐酸,微微加热至透明,冷却后,再用去离子水稀释到100ml。
(8)自配标准汞溶液
a.贮存标准汞溶液:准确称取氯化高汞(分析纯)0.1354g溶于硝酸重铬酸钾溶液中并稀释至 100毫升,即为 1毫克汞/毫升的标准溶液。
b.工作标准溶液 Ⅰ,取 1毫克汞/毫升浓度的标准液 1毫升加硝酸重铬酸钾溶液至 100毫升,即为10.0微克汞/毫升标准溶液。
c.工作标准溶液 Ⅱ,取 10微克汞/毫升标准液 1毫升加硝酸重铬酸钾溶液至 100毫升,即得到0.1微克汞/毫升标准液。
d.绘制工作曲线的标准液,取0.1微克汞/毫升标准液10毫升加硝酸重铬酸钾溶液至 100毫升,得到0.01微克汞/毫升标准液(10ng/ml)。
三、实验步骤
1、 标准曲线绘制
在20ml还原瓶内,分别加入汞浓度为10ng/ml的标准溶液0.00、1.00、2.00、3.00、4.00、5.00ml,再补加硝酸重铬酸钾溶液到液体总体积为5ml,加入1ml氯化亚锡溶液,在仪器上测定它们的吸光度。它们对应的浓度分别为0、2、4、6、8、10微克/升,可在仪器计算直线回归方程和计算标准差、变异系数时使用。
2、 样品测定
取自来水或较清洁废水5ml,加入1ml氯化亚锡溶液,在仪器上测定它的吸光度。在标准曲线上查得其浓度。
思考题:
1、 配制硝酸重铬酸钾溶液的目的是什么?在实验中有什么作用?
2、 实验中为什么要使用氯化亚锡溶液?
实验二 紫外分光光度法测定水中的NO3-—N
一、 实验目的
1、 巩固紫外--可见分光光度法理论知识。
2、 掌握国产紫外分光光度计的基本构成及使用方法。
3、 掌握废水中NO3-—N的紫外分光光度法测定方法
二、 概述
1、 方法原理
利用硝酸根离子在220nm波长处的吸收而定量测定硝酸盐氮。溶解的有机物在220nm处也会有吸收,而硝酸根离子在275nm处没有吸收。因此,在275nm处作另一次测量,以校正硝酸盐氮值。
因此,硝酸根离子在220nm波长处的校正吸收值为 △A = A220 – 2 A275 。
2、干扰及消除
溶解的有机物、表面活性剂、亚硝酸盐、六价铬、溴化物、碳酸氢盐和碳酸盐等干扰测定,需进行适当的预处理。本法采用絮凝共沉淀和大孔中性吸附树脂进行处理,以排除水样中大部分常见有机物、浊度和 Fe3+、Cr6+对测定的干扰。
3、方法的适用范围
本法适用于清洁地表水和未受明显污染的地下水中硝酸盐氮的测定,其最低检出浓度为0.08mg/L,测量上限为 4mg/L硝酸盐氮。
4、仪器
①紫外分光光度计。
②离子交换柱(φ1.4cm,装树脂高5~8cm)。
5、试剂
① 氢氧化铝悬浮液:
②10%硫酸锌溶液。
③5mol/L氢氧化钠溶液。
④大孔径中性树脂:CAD-40或XAD-2型及类似性能的树脂。
⑤甲醇。
⑥lmol/L盐酸(优级纯)。
⑦硝酸盐标准贮备液:每毫升含0.100mg硝酸盐氮。⑧0.8%氨基磺酸溶液:避光保存于冰箱中。
三、 实验步骤
1、吸附柱的制备
新的树脂先用200ml水分两次洗涤,用甲醇浸泡过夜,弃去甲醇,再用40ml甲醇分两次洗涤,然后用新鲜去离子水洗到柱中流出液滴落于烧杯中无乳白色为止。树脂装入柱中时,树脂间绝不允许存在气泡。
2、水样的测定
①量取200ml水样置于锥形瓶或烧杯中,加入2ml硫酸锌溶液,在搅拌下滴加氢氧化钠溶液,调至pH7。或将200ml水样调至pH7后,加 4ml氢氧化铝悬浮液。待絮凝胶团下沉后,或经离心分离,吸取 100ml上清液分两次洗涤吸附树脂柱,以每秒1至2滴的流速流出(注意各个样品间流速保持一致)。弃去。再继续使水样上清液通过柱子,收集50ml于比色管中,备测定用。树脂用150ml水分三次洗涤,备用。
注:树脂吸附容量较大,可处理50~100个地表水水样(视有机物含量而异)。使用多次后,可用未接触过橡胶制品的新鲜去离于水做参比,在220nm和275nm波长处检验,测得的吸光度应接近零。超过仪器允许误差时,需以甲醇再生。
②加 1.0ml盐酸溶液,0.1ml氨基磺酸溶液于比色管中(如亚硝酸盐氮低于0.1mg/L时,可不加氨基磺酸溶液)。
③用光程长10mm石英比色皿,在 220nm和 275nm波长处,以经过树脂吸附的新鲜去离子水50ml加 lml盐酸溶液为参比,测量吸光度。
3、校准曲线的绘制
于6个200ml容量瓶中分别加入0.50、l.00、2.00、3.00、4.00ml硝酸盐氮标准贮备液,用新鲜去离子水稀释至标线,其浓度分别为0.25、0.50、l.00、l.50、2.00mg/L硝酸盐氮。按水样测定相同操作步骤测量吸光度。
四、计算
△A = A220 – 2 A275
式中:A220-220nm波长测得吸光度;
A275-275nm波长测得吸光度。
求得吸光度的校正值(△A )以后,从校准曲线中查得相应的硝酸盐氮量,即为水样定结果(mg/L)。水样若经稀释后测定,则结果应乘以稀释倍数。
五、注意事项
①为了解水受污染程度和变化情况,需对水样进行紫外吸收光谱分布曲线的扫描,如无扫描装置时,可用手动在220~280nm、每隔2~5nm测量吸光度,绘制波长一吸光度曲线。水样与近似浓度的标准溶液分布曲线应类似,且在220nm与275nm附近不应有肩状或折线出现。
参考吸光度比值(A275/A220)* 100%应小于 20%,越小越好。超过时应予以鉴别。
水样经上述方法适用情况检验后,符合要求时,可不经预处理,直接取50ml水样于比色管中,加盐酸和氨基磺酸溶液后,进行吸光度测量。如经絮凝后水样亦达到上述要求,则也可只进行絮凝预处理,省略树脂吸附操作。
②含有有机物的水样,而且硝酸盐含量较高时,必须先进行预处理后再稀释。
③大孔中性吸附树脂对环状、空间结构大的有机物吸附能力强;对低碳链、有较强极性和亲水性的有机物吸附能力差。
④当水样存在六价铬时,絮凝剂应采用氢氧化铝,并放置0.5h以上再取上清液供测定用。
思考题:
1、 在光度分析中参比溶液的作用是什么?
2、 在实验中为什么需要测定校正吸光度?校正吸光度是如何测定的?
实验三 单扫描极谱法测定镉
一、 实验目的
1、 巩固极谱分析理论知识。
2、 掌握国产LK98BⅡ型微机电分析系统的基本构成及使用方法。
3、 掌握废水中镉的单扫描极谱测定方法。
二、 概述
1、 方法原理
普通极谱是在滴汞电极上加一个缓慢变化的电压(每分钟0.2V), 并在一次电压的变化中记录下相应的电流变化,构成所谓极谱电流-电压图。
单扫描极谱基本原理与普通极谱相似, 但是电压扫描的速度比普通极谱快,其在一滴汞的生长后期加一个变化很快的电压到电解池,即扫描电压如三角波,锯齿波或正弦波, 由于电压变化比普通极谱快几十倍,一般为0.25伏/秒,因此引起电流等参数的快速变化,电流- 电位(i-E)曲线可在一滴汞上获得。 在扫描开始时,极化电极的电位还未负到去极剂还原的电位, 此时电流为残余电流,形成极谱波的基线,当电位负到去极剂还原电位时, 电位以很快速度变负,汞滴表面附近去极剂迅速还原,产生很大的电流。随后,电位继续变负,一方面电极表面附近去极剂浓度贫乏;另一方面电解时间增加,增大了扩散层厚度, 从而使电解电流下降,使示波极谱曲线呈峰形。
单扫描极谱的峰值电流与电极反应物质的浓度和扫描速度成正比, 对于可逆极谱波,有:
ip=2.75*105*n3/2*D1/2*V1/2*A*C
其中:n--反应电子数;
D--扩散系数(cm2/sec)
V--扫描速度(V/sec)
A--电极面积(cm2)
C--浓度(mol·L-1)
峰值电位与极谱半波电位的关系:
Ep=E1/2-1.1RT/nF
2、仪器与试剂:
LK98B型微机电分析系统或类似仪器;三电极系统(滴汞电极、铂电极、饱和甘汞电极);极谱工作台。
分析纯Na2SO3、分析纯试剂CdCl2、NH4Cl、NH3· H2O、纯金属Cd、 模似水样(自来水配制)
三、实验步骤:
1.溶液配制:
准确称取纯镉0.100g放入200ml烧怀中,加10ml1:1硝酸溶液,蒸发至干。 加入浓HCl 5ml溶解,移入1000ml容量瓶中,用水稀释到刻度,摇匀。
分别取镉标准溶液0、1、2、3、4ml加入50ml容量瓶中,加入10ml0.1mol·L-1NH4Cl- NH3· H2O 后,用水稀释到刻度,此时溶液中镉的浓度为0、2、4、6、8μg/ml。
取25ml模似水样50ml容量瓶中,加入10ml0.1mol·L-1NH4Cl- NH3· H2O 后,用水稀释到刻度。
2.仪器的启动及自检
①将主机与计算机、与外设以及其他必要设备电源线、控制线连接好。
②打开计算机的电源开关,在Windows98操作平台下运行“LK98BⅡ”,进入主控菜单。
③打开主机的电源开关,按下主机前面板的“RESET”键,这时主控菜单上应显示“系统自检通过”,系统进入正常工作状态。(注意:打开仪器之前请将四根电极线断开,并保证其两两不相连。)
3.实验方法选择及仪器运行参数设定
在主控菜单上选择“线性扫描技术”中的“线性扫描伏安法”,参数设定为:
灵敏度: 10mA 初始电位:- 0.500V
滤波参数:50Hz 终止电位:- 1.000V
放大倍率:1 扫描速度 :0.25V/s
极谱模式:还原电流(+) 等待时间:0s
汞滴生长时间:5s
4.将配制好的各个镉标准溶液及水样分别取适量放入20ml小烧怀中,加入一药勺无水Na2SO3,搅拌溶液片刻,将蓄汞瓶抬升到合适高度,将三电极系统连接到LK主机上,并置于极谱工作台,滴汞电极为阴极、铂电极为阳极,打开极谱工作台电源,将三电极浸入溶液中,按实验“开始”键开始实验,获得电流—电压曲线,于此曲线读出峰电流值及峰电位数据,重复测量俩到叁次,每个溶液读数取一平均值。
5.关机:
测定后冲洗电极,将电极取出后关机。
四、数据处理
将以上各标准溶液所测量峰电流值作电流—浓度校正曲线图。用Excel软件得出此曲线图及校正曲线方程式。
将水样的峰电流值,从校正曲线图上,计算出其浓度,并最终得到模似水样中镉的浓度值。
思考题:
1、单扫描极谱法测定微量金属含量,为什么比经典的极谱法有更高的灵敏度?
2、本实验中为什么要加入Na2SO3?
实验四、阳级溶出伏安法测定水中铜、镉含量
一、 实验目的
1、 掌握阳级溶出伏安法的基本原理。
2、 掌握国产LK98BⅡ型微机电分析系统的基本构成及使用方法。
3、 掌握废水中铜、镉的阳极溶出伏安法的测定方法。
二、 概述
1、 方法原理
溶出伏安法包括阳极溶出伏安法和阴极溶出伏安法。在阳极溶出伏安法中,待测组分在恒定电位下经过富集,金属电沉积在工作电极上,随后,电极电位由负电位向正电位方向快速扫描达到一定电位时,电积的金属经氧化重新以离子状态进入溶液,在这一过程中形成相当强的氧化电流峰。在一定的实验条件下,电流的峰值与待测组分的浓度成正比,借此可进行对该组分的定量分析。
通常以汞膜电极为工作电极,采用非化学计量的电积法,即无须使溶液中全部待测离子电积在工作电极上,这样可缩短电积时间,提高分析速度。为使电积部分的量与溶液中的总量之间维持恒定的比例关系,实验中电积时间、静止时间、扫描速率、电极的位置和搅拌状况等,都应保持严格相同。
设电积时的电流iD很小,在较长的电积时间tD内,可以认为iD不变,则流过的电量为
QD = iD·tD
如果电积的金属在溶出阶段能全部溶出,其流过的电量应与QD相等,并且
QS = iS·tS
式中iS为溶出时的平均电流,tS为溶出时间。采用快速电位扫描技术,可使溶出时间tS大为减少,从而使溶出电流iS相应大为提高。待测组分经过预先的富集,在溶出时突然氧化,使检测信号(溶出峰电流)显著增加,因此溶出伏安法具有较高的灵敏度。
商品化的溶出伏安仪均采用自动控制阴极(或阳极)电位的三电极系统,即除了工作电极(如汞膜电极)和参比电极(如饱和电极)外,增加一个对电极(常用铂电极),以稳定工作电极的电位。
用标准曲线法或标准加入法均可进行定量测定。标准加入法的计算公式为:

式中.Cx、Vx、hx分别为试样的浓度、体积和溶出峰的峰高;
Cs、Vs分别为加入标准溶液的浓度和体积;
H为加人标准溶液后,测得的溶出峰的峰高。
由于加入的标准溶液体积Vs非常小,也可简化为下式计算
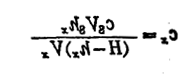
本实验以NH3· H2O-NH4Cl为支持电解电解质,用标准加入法测定水样中Cd2+和Cu2+离子的含量。
2、 仪器
LK98B型微机电分析系统或类似仪器;三电极系统(玻碳汞电极、铂电极、饱和甘汞电极);磁力搅拌器。
3、试剂
(1)10ug/mL的Cd2+、Cu2+离子标准溶液
(2)1mol·L-1NH3· H2O-NH4Cl溶液(pH~10.0)
(3)2*10-2mol·L-1 HgSO4溶液
(4)20% Na2SO3溶液
(5)试样(约含0.2ug/mL Cd2+、Cu2+离子)
三、 实验步骤
1、仪器的启动及自检
①将主机与计算机、与外设以及其他必要设备电源线、控制线连接好。
②打开计算机的电源开关,在Windows98操作平台下运行“LK98BⅡ”,进入主控菜单。
③打开主机的电源开关,按下主机前面板的“RESET”键,这时主控菜单上应显示“系统自检通过”,系统进入正常工作状态。(注意:打开仪器之前请将四根电极线断开,并保证其两两不相连。)
2、于电解杯中加人25mL二次蒸馏水和数滴HgSO4溶液,将玻碳电极抛光洗净后浸入溶液中,在主控菜单上选择“电位阶跃技术”中的“单电位阶跃法”,参数设定为:
灵敏度: 10mA 初始电位:- 0.100V
滤波参数:50Hz 终止电位:- 1.000V
放大倍率:1 采样间隔:0.100s
采样次数:6000
以玻碳电极为阴极,铂电极为阳极, 加入搅拌子,将溶液置于电磁搅拌器上,按实验“开始”键,电镀10min。即得玻碳汞膜电极。
3、于电解杯中加人10mL水样、10mL NH3· H2O-NH4Cl溶液、10mL 20% Na2SO3溶液,加入加入搅拌子,将溶液置于电磁搅拌器上,浸入汞膜电极、铂电极及饱和甘汞电极,将三电极系统连接到LK主机上,在主控菜单上选择“线性扫描技术”中的“线性扫描溶出伏安法”,参数设定为:
灵敏度: 10mA 初始电位:- 0.2000V
滤波参数:50Hz 电积电位:- 1.000V
终止电位:- 0.2000V
放大倍率:1 扫描速度 :0.1000V/s
还原电流(+) 等待时间:0s
清洗电极:无 电积时间:60s
平衡时间:20s
按实验“开始”键开始实验,获得电流—电压曲线,于此曲线读出铜与镉的峰电流值及峰电位数据,重复测量俩到叁次,中间在终止电位上搅拌片刻,以清洗电极。读数取一平均值。在此溶液中加入1mL10ug/mL的Cd2+、Cu2+离子标准溶液,搅拌片刻,重复以上测量。
4、做好实验的结束清理工作。
四、数据及处理
1、记录实验条件
(1)短路清洗时间
(2)电积电位与时间
(3)静止时间
(4)溶出电位范围
2.记录实验数据
3.测量溶出伏安曲线上水样Cd2+、Cu2+离子的各个峰高和加入标准溶液后的各个峰高,并分别取平均值。
4、计算水样中Cd2+、Cu2+离子的浓度(mg/L)。
思考题
2、 结合本实验说明阳极溶出伏安法的基本原理。
3、 溶出伏安法为什么有较高的灵敏度?
4、 实验中为什么对各实验条件必须严格保持一致?