实验五 熔点的测定
一、 实验目的
1、了解熔点测定的意义。
2、掌握熔点测定的操作方法。
二、实验原理
熔点是固体有机化合物固液两态在大气压力下达成平衡的温度。纯净的固体有机化合物一般都有固定的熔点,固液两态之间的变化是非常敏锐的,自初熔至全熔(称为熔程)温度不超过0.5-1℃。
加热纯有机化合物,当温度接近其熔点范围时,升温速度随时间变化约为恒定值,此时用加热时间对温度作图(如图1)。
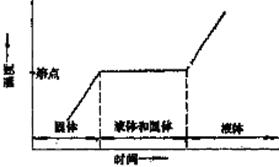

图1 相随时间和温度的变化 图2 物质蒸气压随温度变化曲线
化合物温度不到熔点时以固相存在,加热使温度上升,达到熔点.开始有少量液体出现,而后固液相平衡.继续加热,温度不再变化,此时加热所提供的热量使固相不断转变为液相,两相间仍为平衡,最后的固体熔化后,继续加热则温度线性上升。因此在接近熔点时,加热速度一定要慢,每分钟温度升高不能超过2℃,只有这样,才能使整个熔化过程尽可能接近于两相平衡条件,测得的熔点也越精确。
当含杂质时,根据拉乌耳定律可知,在一定的压力和温度条件下,在溶剂中增加溶质,导致溶剂蒸气分压降低(图2中M´L´),固液两相交点M´即代表含有杂质化合物达到熔点时的固液相平衡共存点,TM´为含杂质时的熔点,显然,此时的熔点较纯粹者低。
三、仪器和药品
四、实验操作
1、样品的装入
将少许样品放于干净表面皿上,用玻璃棒将其研细并集成一堆。把毛细管开口一端垂直插人堆集的样品中,使一些样品进入管内,然后,把该毛细管垂宜桌面轻轻上下振动,使样品进人管底,再用力在桌面上下振动,尽量使样品装得紧密。或将装有样品,管口向上的毛细管,放入长约50一60Cm垂直桌面的玻璃管中,管下可垫一表面皿,使之从高处落于表面皿上,如此反复几次后,可把样品装实,样品高度2—3mm。熔点管外的样品粉末要擦干净以免污染热浴液体。装入的样品一定要研细、夯实。否则影响测定结果。
2、测熔点
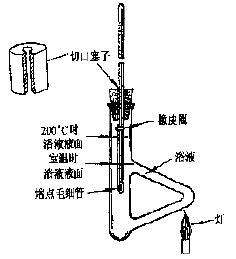
图3熔点测定装置图
按图3搭好装置,放入加热液(液体石腊),剪取一小段橡皮圈套在温度计和熔点管的上部。将粘附有熔点管的温度计小心地插入加热浴中,以小火在图示部位加热。开始时升温速度可以快些,当传热液温度距离该化合物熔点约10一15℃时,调整火焰使每分钟上升约1—2℃,愈接近熔点,升温速度应愈缓慢,每分钟约0.2一0.3℃。为了保证有充分时间让热量由管外传至毛细管内使固体熔化,升温速度是准确测定熔点的关键;另一方面,观察者不可能同时观察温度计所示读数和试样的变化情况,只有缓慢加热才可使此项误差减小。记下试样开始塌落并有液相产生时(初熔)和固体完全消失时(全熔)的温度读数,即为该化合物的熔距。
始熔与全熔的判断:加热过程中,注意观察毛细管内样品的状态变化,将依次出现“发毛”、“收缩”、“液滴”、“澄清”等现象,发毛和收缩以及形成软质柱状物而无液化现象都不是“始熔”,只有当出现液滴(塌落,有液相产生)时才是“始熔”,全部样品变成透明澄清液体时为“全熔”(如图2-13)。记录“始熔”与“全熔”时温度计上所示的温度,即为该化合物的熔程。
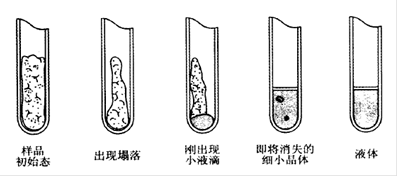
图4 毛细管内样品的状态的变化过程
四、数据记录
第一次测定结果 ;第二次测定结果 。
第二篇:毛细管法测定熔点实验的改进
