
化学与环境学院有机化学设计性实验
题目:以苯胺为原料合成对溴苯胺
姓名:
学号:
专业:
年级:
指导老师:
提交日期:20##年6月6日
以苯胺为原料合成对溴苯胺实验报告
姓名;xxx
(a华南师范大学化学与环境学院 广州 510006)
摘要:对溴苯胺是非常重要的有机化工原料,常被用作染料原料,如偶氮染料、喹啉染料等,医药及有机合成的中间体等。本实验合成过程以苯胺为原料,经历制备乙酰苯胺、对溴乙酰苯胺等中间体的过程,最终制得目标产物对溴苯胺。其合成过程经历保护、溴代、去保护等多个步骤,可以制得纯度较高的对溴苯胺。通过实验可得,用此实验方法制备对溴苯胺,操作方法简单,可控性强,产率较高,此实验最后产率为73.25%。
关键词:有机合成,苯胺,乙酰苯胺,对溴乙酰苯胺,对溴苯胺
The Synthetic Reaction of Aniline to p-Bromoaniline
Abstract: p-Bromoaniline is an important kind of organic chemical material, is often used as dye material, such as the azo dyes, KuiLin dyes, medicine and synthetic organic intermediate, etc. In this experiment, the synthesis process using Aniline as raw material, through preparation Acetyl aniline,Bromine acetyl aniline intermediate, finally makes the product of p-Bromoaniline. The synthesis process through multiple steps to protect, bromination, deprotection, then can be made of high purity of p-Bromoaniline. Through the experiment, with the experimental method of preparing p-Bromoaniline, the method is simple, strong controllability, high yield, the final yied was 73.25%.
Keywords: organic synthesis; Aniline; Acetyl aniline; Bromine acetyl aniline; p-Bromoaniline
引言:溴代苯胺,也被称为4-溴苯胺或对氨基溴化苯,有毒,其毒性较氯苯胺更严重,可经皮肤吸收,具有血溶性、能引起膀胱癌。溴苯胺为无色斜方结晶或粉末,相对分子质量为172.03,熔点为66.4℃,常压沸点分解,相对密度1.4970(100/4℃),对溴苯胺不溶于水,易溶于乙醇和乙醚。
目前,国内生产芳香族胺类多采用酸性条件下铁粉还原法、碱性条件下的硫化碱还原法及中性条件下的Raney-Ni及贵金属还原法。前两种方法虽然成本较低,但对环境污染较为严重,不适合可持续发展的要求。最后一种方案而贵金属( 如Pt、Pd、Ru等) 型催化还原法所用的催化剂价格较贵, 成本高, 虽然回收后可适当降低成本, 但工艺复杂, 目前还不适合工业化生产。
实验室常用的方法是在酸性溶液中用金属进行化学还原,最常用的方法是用锡-盐酸来还原简单的硝基化合物,也可以用铁-醋酸法。苯胺很容易进行酰基化反应,即氨基中的氢原子被酰基取代,这样可以保护芳胺的氨基。卤素对苯环上的取代反应属亲电取代反应,苯环上如有取代基,则按取代基的亲电取代定为规则进行反应。对溴乙酰苯胺在酸性条件下去保护生成目标产物。
本实验总的反应如下:






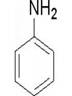
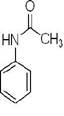
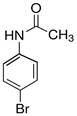
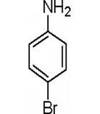
1.实验部分
1.1 实验仪器及药品
1.1.1实验仪器
圆底烧瓶,刺形分馏柱,温度计,量筒,酒精灯,烧杯,玻璃棒,吸滤瓶,布氏漏斗,小水泵,250mL三口烧瓶,电动搅拌器,恒压滴液漏斗,水浴锅,回流冷凝管,蒸馏装置,PH试纸
1.1.2实验药品
苯胺,冰醋酸,锌粉,活性炭,液溴,乙醇,亚硫酸氢钠,浓盐酸,20%氢氧化钠溶液,
1.2实验参数

1.3实验装置图
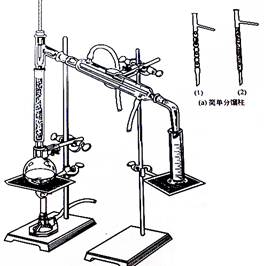
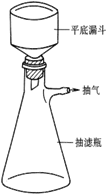
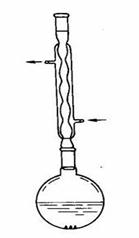
装置1:分馏装置 装置2:抽滤装置 装置3:回流装置

装置4:搅拌滴加回流反应器 装置5: 蒸馏装置
1.4合成
1.4.1 乙酰苯胺的合成




在50 mL 圆底烧瓶中放置10mL苯胺(10.2 g,0.11 mol)、15 mL 冰醋酸(15.7 g,0.26 mol)及少许锌粉(约0.1 g),装上一分馏柱,插上温度计,用小量筒收集蒸出的水和乙酸。将圆底烧瓶在石棉网上小火加热回流,保持温度计读数于105 ℃约1 h,当温度下降则表明反应已经完成。在搅拌下趁热将反应物倒入盛有100 mL 冷水的烧杯中,冷却后抽滤,用冷水洗涤粗产品。将粗产品和水混合,加热溶解,稍冷后,用0.5 g活性炭处理,并煮沸10min。趁热抽滤除去活性炭,滤液冷却,乙酰苯胺结晶析出,抽滤,晾干称重计算产率。
1.4.2 对溴乙酰苯胺的合成


将3.50 g 乙酰苯胺溶解于15.0 mL 乙醇后转移至在250 mL三口烧瓶,配置电动搅拌器、温度计和恒压滴液漏斗,并在恒压滴液漏斗上连接气体吸收装置。将1.31 g(1.5mL) 溴溶解于3 mL 冰醋酸中,一边搅拌一边慢慢地将溴-冰醋酸溶液滴加至乙酰苯胺的醇溶液中,滴加速度以棕红色的溴色较快褪去为宜。滴加完毕,在45 ℃浴温下,继续搅拌反应1 h,然后将浴温提高至60 ℃,再搅拌一段时间,直到反应混合物液面不再有红棕色蒸气溢出为止。在搅拌下将反应物慢慢加至100 mL冷水中,此时即有固体析出。若有黄色,可加入饱和的亚硫酸氢钠水溶液洗涤,使溶液黄色恰好褪去。减压过滤收集产物,并用冷水充分洗涤,干燥,用乙醇重结晶,得到白色针状晶体。
1.4.3 对溴苯胺的合成



安装回流反应装置,向三口烧瓶中加入上面得到的3.9g对溴乙酰苯胺、10mL95%的乙醇和三粒沸石,加热至沸,自恒压滴液漏斗缓慢滴加4.5mL的浓盐酸。加毕回流30分钟,加入20mL水使反应物稀释。将回流装置改成蒸馏装置进行蒸馏,将残余物对溴苯胺盐倒入30mL冰水中,在搅拌下滴加20%氢氧化钠使之刚好呈碱性。抽滤,水洗抽干后用乙醇-水重结晶,自然晾干,称重计算产率。
2.结果与讨论
2.1产物产率
对溴苯胺的全合成各步骤制备产率见表1
表1 对溴苯胺全合成实验数据
Table1 The experiment data about total synthetic of p-bromoaniline

2.2讨论
2.2.1乙酰苯胺的制备
a. 为什么要控制温度在105℃左右?且在反应终点时,为什么温度会下降?
乙酰苯胺的沸点为114℃,冰乙酸的沸点为117.9℃,若反应温度超过乙酰苯胺和冰乙酸的沸点,则反应物会大量挥发,且苯胺高温不稳定,易被氧化,因此要保持温度在105℃左右。在到达反应终点时,生成的水已大部分蒸干,但并未达到乙酰苯胺的沸点,因此温度会下降。
b. 如何提高乙酰苯胺的产率?
冰乙酸与苯胺的反应速率较慢,且反应可逆,因此可通过冰乙酸过量的方式提高产率。
利用分馏柱将反应中生成的水从反应平衡中移去,使得反应向生成乙酰苯胺的方向进行。
加入锌粉防止苯胺被氧化,减少反应物的损失。
2.2.2 对溴乙酰苯胺的制备
对溴乙酰苯胺的制备中,滴溴速度对产物生成的影响很大。滴溴速度不宜过快,否则会导致一部分溴来不及参与反应就与溴化氢一起逸出,同时也可能会产生二溴代产物。本次实验滴溴速度不快,且控制好滴加的量,对产率影响不大。但由于溴的量过量,使得溶液的黄色即使在水浴加热时也没有退去,且用亚硫酸氢钠除色时未除干净导致产品颜色有差异。
2.2.3 对溴苯胺的制备
如何提高目标产物的产率?
对粗产品重结晶时应用尽量少的乙醇-水,否则产物会损失较多。
用对溴乙酰苯胺制备目标产物时要用小火加热以防生成的对溴苯胺再次被氧化而造成目标产物的损失,影响产率。
2.2.4 酸性的选择
本实验的每一个步骤都用到酸,因此,酸性的选择对实验具有重要作用。
【1】中间体乙酰苯胺的制备时,常用的乙酰化试剂有乙酰氯、乙酸酐和冰乙酸,乙酰氯的反应活性最大,但价格昂贵,因此应用不广;乙酸酐一般来说是较好的酰基化试剂若在HAc-NaAc缓冲溶液中反应,其水解速率比酰化速率慢,因此能得到高纯产物,价格较贵;而冰乙酸成本最低,乙酰化效果好,但是反应时间长,适合于大规模的制备;为了降低成本,提高产率,本制备采用过量的冰乙酸为酰化试剂。
【2】利用亲电取代反应:由乙酰苯胺制备对溴乙酰苯胺时,酸性增加了溴的亲电性。
【3】对溴乙酰苯胺在酸性条件下去保护生成对溴苯胺,但浓盐酸滴加速度不宜太快,否则反应太剧烈导致部分浓盐酸挥发,反应不充分。
2.3 结论
本实验合成过程以苯胺为原料,经历制备乙酰苯胺、对溴乙酰苯胺等中间体的过程,最终制得目标产物对溴苯胺。其合成过程经历保护、溴代、去保护等多个步骤,可以制得纯度较高的对溴苯胺,中间体的产率直接影响目标产物的合成。通过实验可得,本实验可以通过提高重结晶产率来提高目标产物的产率,用此实验方法制备对溴苯胺,操作方法简单,可控性强,产率较高,此实验最后产率为73.25%。
参考文献:
[1] 杨定乔. 对溴苯胺的全合成 [M] .有机化学实验,163-169
[2] 鲁莉华,李英春.催化氢化法制备对溴苯胺[J].青岛科技大学学报,20##,26(3):025-027
[3] 李广洲,徐永进.苯胺的制备.化学教学,1996(1)
[4] 李康兰,张力超声波辐射促进对溴苯胺的合成[J]. 甘肃省化学会第二十五届年会、第七届甘肃省中学化学教学经验交流会论文集,20##; O625.63
[5] 张洪权,郑平,田露.4-溴代乙酰苯胺制备的半微型实验研究.黑龙江医药科学,20##(4)
[6] Padeya SN, Yogeeswari PD, Sausville EA. Synthesis an dantitumor evaluation of 4-bromo phenyl semi carbaz ones [J] .Arzneimittel-Forschung, 20##, 52(2): 103-108.
[7] Isobe Koji;Nakano Yasuo;Fujise Masatomo Halogenated aromatic primary amines from halo nitro compounds and a platinum catalyst 1973
第二篇:有机化学实验报告 (有课后题)
实验一 玻璃工操作和塞子的钻孔
一、实验目的及基本要求
在有机化学实验中,常常需要多种不同角度的弯管,直径不同的毛细管及滴管和搅拌棒等用品,因此必须掌握一些简单的玻璃工操作,才能自己动手制这些用品。
通过实验,要求学生能够掌握简单玻璃工操作的基本要领,会弯不同角度的玻璃弯管,会拉不同直径的毛细管和制电动搅拌器上的玻璃搅拌棒等。
二、操作要点和说明
1、玻璃弯管的制备
有机化学实验中常用的玻璃弯管有45o、75o、90o、135o等。初学者容易出现的问题有:弯曲部分变细了,扭曲了,瘪了等。为此:
(1)加热部分要稍宽些,同时要不时转动使其受热均匀。
(2)不能一面加热一面弯曲,一定要等玻璃管烧软后离开火焰再弯,弯曲时两手用力要均匀,不能有扭力,拉力和推力。
(3)玻璃管弯曲角度较大时,不能一次弯成,先弯曲一定角度将加热中心部位稍偏离原中心部位,再加热弯曲,直至达到所要求的角度为止。
(4)弯制好的玻璃弯管不能立即和冷的物件接触,要把它放在石棉网(板)上自然冷却。
(5)水蒸汽导人管的弯制。见曾绍琼书P54。
2、毛细管的拉制
有机化学实验中常用的毛细管有熔点管、沸点管、薄等层层析点样用的毛细管,减压蒸馏用的毛细管及滴管等,内径要求各不相同。初学者容易出现的问题及克服方法是:
(1)玻璃管尚未烧柔软就拉,把玻璃管拉成了亚铃形,所以一定要等玻璃管烧软化后再拉,软化程度要比弯玻璃管强一些。
(2)玻璃管尚未离开火焰就拉,毛细管很快被拉断,所以要等玻璃管烧软化后离开火焰再拉,拉的速度既不能太快也不能大慢。应根据毛细管内径要求而定,内径小的可快点,内径大的可慢点。
(3)拉后的毛细管未等冷却就立即放在台子上,至使毛细管两端弯曲或破裂,所以拉毛细管时,两手要端平,使玻璃管烧软化后离开火焰向相反方向拉,拉后稍停片刻再放到垫有石棉网(板)的台子上冷却。
3、玻璃搅拌棒的制备
这里所说的玻璃搅拌棒,是指装在电动搅拌头上的搅拌棒。这里介绍一种制备简单,搅拌效果又好的玻璃搅拌棒的制法。
取一根一定长度的玻璃棒,在煤气灯火焰上将距一端约2cm处烧软后,先弯成135℃,再将弯曲部分烧软化后放在石棉网(板)上,用老虎钳等硬物压扁即可.
三、思考题
1、弯曲和拉细玻璃管时,玻璃管的温度有什么不同?为什么要不同呢?弯制好了的玻璃管,如果和冷的物件接触会发生什么不良的后果?应该怎样才能避免?
答:拉制玻璃管时,玻璃管的温度比弯曲玻璃管时玻璃管温度要稍高,玻璃管的软化程度要强些,否则拉不动(细)。弯制好了的曲玻璃管,如果和冷的物件按触,玻璃管很容易破裂,为此可将弯制好了的曲玻璃管放在石棉网(板)上自然冷却。
2、在强热玻离管(棒)之前,应用小火加热。在加工完毕后又需小火“退火”,这是为什么?
答:在强热之前先用小火加热,使玻璃管(棒)的温度逐渐升高,否则如果直接强热,温度变化太快将会引起玻离管(棒)破裂。加工完毕后,小火“退火”,也是为使温度逐渐降低,否则,如温度突然降低,也会使玻璃管(棒)发生破裂。
3、把玻璃管插入塞子孔道中时要注意些什么?怎样才不会割破皮肤呢?拔出时要怎样操作才安全?
答:把玻璃管插入塞子孔道中时要注意的是:(1)孔道直径要适宜;(2)管端可蘸点水或甘油或凡士林润滑;(3)握玻离管的手离插入端尽量近些以减小扭矩;(4)要旋转前进,不要硬推,做到这些就不会割破皮肤了。
拔出时要注意的是:(1)握玻璃管的手离插入端尽量近,也为减小扭矩。(2)一面旋转一面用力拔,而不能只用力拔而不旋转。(3)对长时间插入橡皮塞的玻璃管一般较难拔掉,这是因为橡胶老化使其与玻璃管粘牢的缘故,遇到这种情况只有用刀片切开拿掉了。
实验二 熔点的测定和温度计刻度的校正
一、实验目的及基本要求
物质熔点的测定是有机化学工作者经常用的一种技术,所得的数据可用来鉴定晶状的有机化合物,并作为该化合物纯度的一种指标。
目前测熔点的方法有:(1)毛细管法测熔点; (2)熔点测定仪测熔点。
通过实验主要是要让学生掌握用毛细管法测定固体有机物质熔点的操作方法,了解显微熔点测定法原理及操作技术。
二、基本原理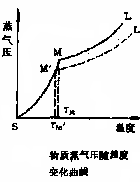
物质的温度与蒸气压的关系如下图所示
曲线SM——物质固相的蒸气压与温度的关系
曲线ML——物质液相的蒸气压与温度的关系(因为两条线斜率不同,所以有交点)
M——固液两相蒸气压一致,固液两相平衡共存,这时的温度TM即为该物质的熔点。
三、操作要点和说明
影响毛细管法测熔点的主要因素及措施有:
1、熔点管本身要干净,管壁不能太厚,封口要均匀。初学者容易出现的问题是,
封口一端发生弯曲和封口端壁太厚,所以在毛细管封口时,一端在火焰上加热时要尽量让毛细管接近垂直方向,火焰温度不宜太高,最好用酒精灯,断断续续地加热,封口要圆滑,以不漏气为原则。
2、样品一定要干燥,并要研成细粉末,往毛细管内装样品时,一定要反复冲撞夯实,管外样品要用卫生纸擦干净。
3、用橡皮圈将毛细管缚在温度计旁,并使装样部分和温度计水银球处在同一水平位置,同时要使温度计水银球处于b形管两侧管中心部位。
4、升温速度不宜太快,特别是当温度将要接近该样品的熔点时,升温速度更不能快。一般情况是,开始升温时速度可稍快些(5℃/min)但接近该样品熔点时,升温速度要慢(1-2℃/min),对未知物熔点的测定,第一次可快速升温,测定化合物的大概熔点。
5、熔点温度范围(熔程、熔点、熔距)的观察和记录,注意观察时,样品开始萎缩(蹋落)并非熔化开始的指示信号,实际的熔化开始于能看到第一滴液体时,记下此时的温度,到所有晶体完全消失呈透明液体时再记下这时的温度,这两个温度即为该样品的熔点范围。
6、熔点的测定至少要有两次重复的数据,每一次测定都必须用新的熔点管,装新样品。进行第二次测定时,要等浴温冷至其熔点以下约30℃左右再进行。
7、使用硫酸作加热浴液(加热介质)要特别小心,不能让有机物碰到浓硫酸,否则使溶液颜色变深,有碍熔点的观察。若出现这种情况,可加人少许硝酸钾晶体共热后使之脱色。采用浓硫酸作热浴,适用于测熔点在220℃以下的样品。若要测熔点在220℃以上的样品可用其它热浴液。见兰大、复旦书P431。
8、测定工作结束,一定要等浴液冷却后方可将浓硫酸倒回瓶中。温度计也要等冷却后,用废纸擦去硫酸方可用水冲洗,否则温度计极易炸裂。
9、用熔点仪测熔点见北师大《化学实验规范》 P256。
四、思考题
1、测熔点时,若有下列情况将产生什么结果?
(1)熔点管壁太厚。
(2)熔点管底部未完全封闭,尚有一针孔。
(3)熔点管不洁净。
(4)样品未完全干燥或含有杂质。
(5)样品研得不细或装得不紧密。
(6)加热太快。
答:(1)管壁太厚样品受热不均匀,熔点测不准,熔点数据易偏高,熔程大。
(2)熔点管底部未完全封闭有一针孔,空气会进人,加热时,可看到有气泡从溶液中跑出接着溶液进人,结晶很快熔化,也测不准,偏低。
(3)熔点管不洁净,等于样品中有杂质,致使测得熔点偏低,熔程加大。
(4)样品未完全干燥,内有水分和其它溶剂,加热,溶剂气化,使样品松动熔化,也使所测熔点数据偏低,熔程加大.样品含有杂质的话情况同上。
(5)样品研得不细和装得不紧密,里面含有空隙,充满空气,而空气导热系数小传热慢,会使所测熔点数据偏高熔程大。
(6)加热太快,升温大快,会使所测熔点数据偏高,熔程大,所以加热不能太快。这一方面是为了保证有充分的时间让热量由管外传至管内,以使固体熔化。另一方面因观察者不能同时观察温度计所示度数和样品的变化情况,只有缓慢加热才能使此项误差变小。
2、是否可以使用第一次测过熔点时已经熔化的有机化合物再作第二次测定呢?为什么?
答:不可以,这是因为第一次测过熔点后,有时有些物质会产生部分分解,有些会转变成具有不同熔点的其它结晶形式。
实验三 蒸馏、分馏和沸点的测定
一、实验目的和基本要求
蒸馏和分馏的基本原理是一样的,都是利用有机物质的沸点不同,在蒸馏过程中低沸点的组分先蒸出,高沸点的组分后蒸出,从而达到分离提纯的目的。不同的是,分馏是借助于分馏柱使一系列的蒸馏不需多次重复,一次得以完成的蒸馏(分馏就是多次蒸馏),应用范围也不同,蒸馏时混合液体中各组分的沸点要相差30℃以上,才可以进行分离,而要彻底分离沸点要相差110℃以上。分馏可使沸点相近的互溶液体混合物(甚至沸点仅相差1-2℃)得到分离和纯化。
通过实验使学生:
(1)理解蒸馏和分馏的基本原理,应用范围,什么情况下用蒸馏,什么情况下用分馏。
(2)熟练掌握蒸馏装置的安装和使用方法。
(3)掌握分馏柱的工作原理和常压下的简单分馏操作方法。
二、基本原理
当液态物质受热时蒸气压增大,待蒸气压大到与大气压或所给压力相等时液体沸腾,即达到沸点。所谓蒸馏就是将液态物质加热到沸腾变为蒸气,又将蒸气冷却为液体这两个过程的联合操作。
分馏:如果将两种挥发性液体混合物进行蒸馏,在沸腾温度下,其气相与液相达成平衡,出来的蒸气中含有较多量易挥发物质的组分,将此蒸气冷凝成液体,其组成与气相组成等同(即含有较多的易挥发组分),而残 留物中却含有较多量的高沸点组分(难挥发组分),这就是进行了一次简单的蒸馏。
如果将蒸气凝成的液体重新蒸馏,即又进行一次气液平衡,再度产生的蒸气中,所含的易挥发物质组分又有增高,同样,将此蒸气再经冷凝而得到的液体中,易挥发物质的组成当然更高,这样我们可以利用一连串的有系统的重复蒸馏,最后能得到接近纯组分的两种液体。
应用这样反复多次的简单蒸馏,虽然可以得到接近纯组分的两种液体,但是这样做既浪费时间,且在重复多次蒸馏操作中的损失又很大,设备复杂,所以,通常是利用分馏柱进行多次气化和冷凝,这就是分馏。
在分馏柱内,当上升的蒸气与下降的冷凝液互凝相接触时,上升的蒸气部分冷凝放出热量使下降的冷凝液部分气化,两者之间发生了热量交换,其结果,上升蒸气中易挥发组分增加,而下降的冷凝液中高沸点组分(难挥发组分)增加,如果继续多次,就等于进行了多次的气液平衡,即达到了多次蒸馏的效果。这样靠近分馏柱顶部易挥发物质的组分比率高,而在烧瓶里高沸点组分(难挥发组分)的比率高。这样只要分馏柱足够高,就可将这种组分完全彻底分开。工业上的精馏塔就相当于分馏柱。
三、操作要点和说明
1、进行蒸馏操作时,有时发现馏出物的沸点往往低于(或高于)该化合物的沸点,有时馏出物的温度一直在上升,这可能是因为混合液体组成比较复杂,沸点又比较接近的缘故,简单蒸馏难以将它们分开,可考虑用分馏。
2、沸石的加入 为了清除在蒸馏过程中的过热现象和保证沸腾的平稳状态,常加沸石,或一端封口的毛细管,因为它们都能防止加热时的暴沸现象,,把它们称做止暴剂又叫助沸剂,值得注意的是,不能在液体沸腾时,加入止暴剂,不能用已使用过的止暴剂。
3、蒸馏及分馏效果好坏与操作条件有直接关系,其中最主要的是控制馏出液流出速度,以1-2滴/s为宜(lml/min),不能太快,否则达不到分离要求。
4、当蒸馏沸点高于140℃的物质时,应该使用空气冷凝管。
5、如果维持原来加热程度,不再有馏出液蒸出,温度突然下降时,就应停止蒸馏,即使杂质量很少也不能蒸干,特别是蒸馏低沸点液体时更要注意不能蒸干,否则易发生意外事故。蒸馏完毕,先停止加热,后停止通冷却水,拆卸仪器,其程序和安装时相反。
6、蒸馏低沸点易燃吸潮的液体时,在接液管的支管处,连一于燥管,再从后者出口处接胶管通入水槽或室外,并将接受瓶在冰浴中冷却。
7、简单分馏操作和蒸馏大致相同,要很好地进行分馏,必须注意下列几点:
(1)分馏一定要缓慢进行,控制好恒定的蒸馏速度(1-2/s),这样,可以得到比较好的分馏效果。
(2)要使有相当量的液体沿柱流回烧瓶中,即要选择合适的回流比,使上升的气流和下降液体充分进行热交换,使易挥发组分量上升,难挥发组分尽量下降,分馏效果更好。
(3)必须尽量减少分馏柱的热量损失和波动。柱的外围可用石棉绳包住,这样可以减少柱内热量的散发,减少风和室温的影响也减少了热量的损失和波动,使加热均匀,分馏操作平稳地进行。
四、思考题
1、什么叫沸点?液体的沸点和大气压有什么关系?文献里记载的某物质的沸点是否即为你们那里的沸点温度?
答:将液体加热,其蒸气压增大到和外界施于液面的总压力(通常是大气压力)相等时,液体沸腾,此时的温度即为该液体的沸点。
文献上记载的某物质的沸点不一定即为我们那里的沸点度,通常文献上记载的某物质的沸点,如不加说明,一般是一个大气压时的沸点,如果我们那里的大气压不是一个大气压的话,该液体的沸点会有变化。
2、蒸馏时加入沸石的作用是什么?如果蒸馏前忘记加沸石,能否立即将沸石加至将近沸腾的液体中?当重新蒸馏时,用过的沸石能否继续使用?
答:加入沸石的作用是起助沸作用,防止暴沸,因为沸石表面均有微孔,内有空气,所以可起助沸作用。不能将沸石加至将近沸腾的液体中,那样溶液猛烈暴沸,液体易冲出瓶口,若是易燃液体,还会引起火灾,要等沸腾的液体冷下来再加。
用过的沸石一般不能再继续使用,因为它的微孔中已充满或留有杂质,孔经变小或堵塞,不能再起助沸作用。
3、为什么蒸馏时最好控制馏出液的速度为1-2滴/s为宜?
答:在整个蒸馏过程中,应使温度计水银球上常有被冷凝的液滴,让水银球上液滴和蒸气温度达到平衡。所以要控制加热温度,调节蒸馏速度,通常以1-2滴/s为宜,否则不成平衡。蒸馏时加热的火焰不能太大,否则会在蒸馏瓶的颈部造成过热现象,使一部分液体的蒸气直接受到火焰的热量,这样由温度计读得的沸点会偏高;另一方面,蒸馏也不能进行的太慢,否则由于温度计的水银球不能为馏出液蒸气充分浸润而使温度计上所读得的沸点偏低或不规则。
4、如果液体具有恒定的沸点,那么能否认为它是单纯物质?
答:纯粹的液体有机化合物,在一定的压力下具有一定的沸点,但是具有固定沸点的液体不一定都是纯粹的化合物,因为某些有机化合物常和其它组分形成二元或三元共沸混合物,它们也有一定的沸点。
5、分馏和蒸馏在原理及装置上有哪些异同?如果是两种沸点很接近的液体组成的混合物能否用分馏来提纯呢?
答:利用蒸馏和分馏来分离混合物的原理是一样的,实际上分馏就是多次的蒸馏。分馏是借助于分馏往使一系列的蒸馏不需多次重复。一次得以完成的蒸馏。
现在,最精密的分馏设备已能将沸点相差仅1-2℃混合物分开,所以两种沸点很接近的液体组成的混合物能用分馏来提纯。
6、若加热太快,馏出液>1-2滴/s(每秒种的滴数超过要求量),用分馏分离两种液体的能力会显著下降,为什么?
答:因为加热太快,馏出速度太快,热量来不及交换(易挥发组分和难挥发组分),致使水银球周围液滴和蒸气未达平衡,一部分难挥发组分也被气化上升而冷凝,来不及分离就一道被蒸出,所以分离两种液体的能力会显著下降。
7、用分馏柱提纯液体时,为了取得较好的分离效果,为什么分馏柱必须保持回流液?
答:保持回流液的目的在于让上升的蒸气和回流液体,充分进行热交换,促使易挥发组分上升,难挥发组分下降,从而达到彻底分离它们的目的。
8、在分离两种沸点相近的液体时,为什么装有填料的分馏柱比不装填料的效率高?
答:装有填料的分馏柱上升蒸气和下降液体(回流)之间的接触面加大,更有利于它们充分进行热交换,使易挥发的组分和难挥发组分更好地分开,所以效率比不装填料的要高。
9、什么叫共沸物?为什么不能用分馏法分离共沸混合物?
答:当某两种或三种液体以一定比例混合,可组成具有固定沸点的混合物,将这种混合物加热至沸腾时,在气液平衡体系中,气相组成和液相组成一样,故不能使用分馏法将其分离出来,只能得到按一定比例组成的混合物,这种混合物称为共沸混合物或恒沸混合物。
10、在分馏时通常用水浴或油浴加热,它比直接火加热有什么优点?
答:在分馏时通常用水浴或油浴,使液体受热均匀,不易产生局部过热,这比直接火加热要好得多。
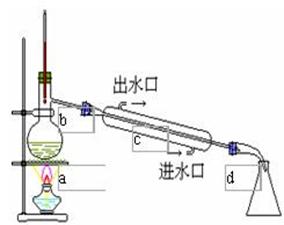
实验四 水蒸汽蒸馏
一、实验目的和基本要求
水蒸汽蒸馏操作是将水蒸气通入不溶或难溶于水,但有一定挥发度(性)的有机物质(进100℃其蒸气压至少为5-10mmHg,665.5—1333Pa)中,使该有机物质在低于100℃的温度下,随着水蒸汽一起蒸馏出来。
通过实验使学生:
(1)了解水蒸汽蒸馏的基本原理,使用范围(场合)和被蒸馏物应具备的条件。
(2)熟练掌握常量水蒸气蒸馏仪器的组装和使用方法。
二、基本原理
根据道尔顿分压定律,两种互不相溶的液体混合物的蒸气压等于两液体单独存在时的蒸气压之和。 因为当组成混合物的两液体的蒸气压之和等于大气压力时混合物就开始沸腾(此时的温度为共沸点)。所以互不相溶的液体混合物的沸点,要比每一物质单独存在时的沸点低。
因此,在不溶于水的有机物质中,进行水蒸汽蒸馏时,在比该物质的沸点低得多的温度,而且比100℃还要低的温度就可使该物质和水一起蒸馏出来。
三、操作要点和说明
1、蒸馏烧瓶的容量应保证混合物的体积不超过其1/3,导入蒸汽的玻管下端应垂直地正对瓶底中央,并伸到接近瓶底。安装时要倾斜一定的角度,通常为45℃左右。
2、水蒸汽发生器上的安全管(平衡管)不宜太短,其下端应接近器底,盛水量通常为其容量的1/2,最多不超过2/3,最好在水蒸汽发生器中加进沸石起助沸作用。
3、应尽量缩短水蒸汽发生器与蒸馏烧瓶之间的距离,以减少水汽的冷凝。
4、开始蒸馏前应把T形管上的止水夹打开,当T形管的支管有水蒸汽冲出时,接通冷凝水,开始通水蒸汽,进行蒸馏。
5、为使水蒸汽不致在烧瓶中冷凝过多而增加混合物的体积。在通水蒸汽时,可在烧瓶下用小火加热。
6、在蒸馏过程中,要经常检查安全管中的水位是否正合,如发现其突然升高,意味着有堵塞现象,应立即打开止水夹,移去热源,使水蒸汽发生器与大气相通,避免发生事故(如倒吸),待故障排除后再行蒸馏。如发现T形管支管处水积聚过多,超过支管部分,也应打开止水夹,将水放掉,否则将影响水蒸汽通过。
7、当馏出液澄清透明,不含有油珠状的有机物时,即可停止蒸馏,这时也应首先打开夹子,然后移去热源。
8、如果随水蒸汽挥发馏出的物质熔点较高,在冷凝管中易凝成固体堵塞冷凝管,可考虑改用空气冷凝管。
四、思考题
1、进行水蒸汽蒸馏时,蒸汽导入管的末端为什么要插入到接近于容器的底部?
答:插入容器底部的目的是使瓶内液体充分加热和搅拌,有利于更有效地进行水蒸汽蒸馏。
2、在水蒸汽蒸馏过程中,经常要检查什么事项?若安全管中水位上升很高说明什么问题,如何处理才能解决呢?
答:经常要检查安全管中水位是否正常,若安全管中水位上升很高,说明系统有堵塞现象,这时应立即打开止水夹,让水蒸汽发生器和大气相通,排除故障后方可继续进行蒸馏。
实验五 减压蒸馏(真空蒸馏)
一、实验目的和基本要求
减压蒸馏是分离和提纯高沸点和性质不稳定的液体以及一些低熔点固体有机物的常用方法。
应用这一方法可将沸点高的物质以及在普通蒸馏时还没达到沸点温度就已分解,氧化或聚合的物质纯化。
通过实验让学生
(1)了解减压蒸馏的原理和应用范围。
(2)认识减压蒸馏的主要仪器设备。
(3)掌握减压蒸馏仪器的安装和操作方法。
二、基本原理
已知液体的沸点是指它的蒸气压等于外界大气压时的温度。所以液体沸腾的温度是随外再压力的降低而降低的。因而用真空泵连接盛有液体的容器,使液体表面上的压力降低,即可降低液体的沸点。这种在较低压力下进行蒸馏的操作称为减压蒸馏,减压蒸馏时物质的沸点与压力有关。
三、操作要点和说明
常用的减压蒸馏系统可分为蒸馏、抽气以及保护和测压装置三部分。
1、蒸馏部分
这一部分与普通蒸馏相似,亦可分为三个组成部分
(1)减压蒸馏瓶(克氏蒸馏瓶)有两个颈,其目的是为了避免减压蒸馏时瓶内液体由于沸腾而冲入冷凝管中,瓶的一颈中插入温度计,另一颈中插入一根距瓶底约1-2mm的末端拉成细丝的毛细管的玻管。毛细管的上端连有一段带螺旋夹的橡皮管,螺旋夹用以调节进入空气的量,使极少量的空气进入液体,呈微小气泡冒出,作为液体沸腾的气化中心,使蒸馏平稳进行,又起搅拌作用。
(2)冷凝管和普通蒸馏相同。
(3)接液管(尾接管)和普通蒸馏不同的是,接液管上具有可供接抽气部分的小支管。蒸馏时,若要收集不同的馏分而又不中断蒸馏,则可用两尾或多尾接液管。转动多尾接液管,就可使不同的馏分进入指定的接受器中。
2、抽气部分
实验室通常用水泵或油泵进行减压。
水泵(水循环泵):所能达到的最低压力为0.1Pa
油泵:油泵的效能决定于油泵的机械结构以及真空泵油的好坏。好的油泵能抽至真空度为13.3Pa。油泵结构较精密,工作条件要求较严。蒸馏时,如果有挥发性的有机溶剂、水或酸的蒸气,都会损坏油泵及降低其真空度。因此,使用时必须十分注意油泵的保护。
3、保护和测压装置部分
为了保护油泵必须在馏液接受器与油泵之间顺次安装冷阱和几个吸收塔。冷阱中冷却剂的选择随需要而定。吸职塔(干燥塔)通常设三个:第一个装无水CaCl2或硅胶,吸收水汽;第二个装粒状Na0H,吸酸性气体;第三个装切片石腊,吸烃类气体。
实验室通常利用水银压力计来测量减压系统的压力。水银压力计又有开口式水银压力计、封闭式水银压力计。
三、操作要点及说明
(1)被蒸馏液体中若含有低沸点物质时,通常先进行普通蒸馏,再进行水泵减压蒸馏,而油泵减压蒸馏应在水泵减压蒸馏后进行。
(2)装置停当后,先旋紧橡皮管上的螺旋夹,打开安全瓶上的二通活塞,使体系与大气相通,启动油泵(长时间未用的真空泵,启动前应先用手转动下皮带轮,能转动时再启动)抽气,逐渐关闭二通活塞至完全关闭,注意观察瓶内的鼓泡情况(如发现鼓泡太剧烈,有冲料危险,立即将二通活塞旋开些)从压力计上观察体系内压力应能符合要求,然后小心旋开二通活塞,同时注意观察压力计上的读数,调节体系内压到所需值(根据沸点与压力关系)。
(3)在系统充分抽空后通冷凝水,再加热(一般用油溶)蒸馏,一旦减压蒸馏开始,就应密切注意蒸馏情况,调整体系内压,经常记录压力和相应的沸点值,根据要求,收集不同馏分。
(4)蒸馏完毕,移去热源,慢慢旋开螺旋夹(防止倒吸),并慢慢打开二通活塞,平衡内外压力,使测压计的水银柱慢慢地回复原状,(若打开得太快,水银柱很快上升,有冲破测压计的可能),然后关闭油泵和冷却水。
四、思考题
1、在怎样的情况下才用减压蒸馏?
答:沸点高的物质以及在普通蒸馏时还没达到沸点温度就已分解,氧化或聚合的物质才用减次蒸馏。
2、使用油泵减压时,实有哪些吸收和保护装置?其作用是什么?
答:油泵的结构较精密,工作条件要求较严,蒸馏时如有挥发性的有机溶剂、水、酸蒸气都会损坏泵和改变真空度。所以要有吸收和保护装置。
主要有:(1)冷阱:使低沸点(易挥发)物质冷凝下来不致进入真空泵。(2)无水CaCl2干燥塔,吸收水汽。(3)粒状氢氧化钠塔,吸收酸性气体。(4)切片石腊:吸收烃类物质。
3、在进行减压蒸馏时,为什么必须用热浴加热,而不能用直接火加热?为什么进行减压蒸馏时须先抽气才能加热?
答:用热浴的好处是加热均匀,可防止暴沸,如果直接用火加热的话,情况正好相反。因为系统内充满空气,加热后部分溶液气化,再抽气时,大量气体来不及冷凝和吸收,会直接进入真空泵,损坏泵改变真空度。如先抽气再加热,可以避免或减少之。
4、当减压蒸完所要的化合物后,应如何停止减压蒸馏?为什么?
答:蒸馏完毕移去热源,慢慢旋开螺旋夹,并慢慢打开二通活塞,(这样可以防止倒吸),平衡内外压力,使测压计的水银柱慢慢地回复原状(若放开得太快,水银柱很快上升,有冲破压力计的可能)然后关闭油泵和冷却水。
实验六 重结晶提纯法
一、实验目的和基本要求
重结晶是纯化精制固体有机化合物的手段。
通过实验让学生能熟练掌握用水、有机溶剂及混合溶剂重结晶纯化固体有机物质的各项具体的操作方法,其中包括以下几点:
(1)样品的溶解,突出用易燃的有机溶剂时溶解样品应采用仪器装置及安会注意事项。
(2)过滤及热过滤;菊花滤纸的折法。
(3)结晶及用活性炭脱色。
(4)抽滤:布氏漏斗、抽滤瓶、安全瓶、循环水泵等的安装及使用。
(5)产品的干燥,包括风干(自然晾干)和烘干(使用烘箱、红外干燥)时仪器的使用及注意事项。
二、基本原理
固体有机物在溶剂中的溶解度与温度有密切关系。一般是温度升高, 溶解度增大。利用溶剂对被提纯物质及杂质的溶解度不同,可以使被提纯物质从过饱和溶液中析出,而让杂质全部或大部分仍留在溶液中,或者相反,从而达到分离、提纯之目的。
三、操作要点及说明
重结晶提纯法的一般过程为:
1、选择适宜的溶剂
在选择溶剂时应根据“相似相溶”的一般原理。溶质往往溶于结构与其相似的溶剂中。还可查阅有关的文献和手册,了解某化合物在各种溶剂中不同温度的溶解度。也可通过实验来确定化合物的溶解度。即可取少量的重结晶物质在试管中,加入不同种类的溶剂进行预试。适宜溶剂应符合的条件:见曾绍琼书P61。
2、将待重结晶物质制成热的饱和溶液
制饱和溶液时,溶剂可分批加入,边加热边搅拌,至固体完全溶解后,再多加2O%左右(这样可避免热过滤时,晶体在漏斗上或漏斗颈中析出造成损失)。切不可再多加溶剂,否则冷后析不出晶体。
如需脱色,待溶液稍冷后,加入活性炭(用量为固体1-5%),煮沸5-10min(切不可在沸腾的溶液中加入活性炭,那样会有暴沸的危险。)
3、乘热过滤除去不溶性杂质
乘热过滤时,先熟悉热水漏斗的构造,放入菊花滤纸(要使菊花滤纸向外突出的棱角,紧贴于漏斗壁上),先用少量热的溶剂润湿滤纸(以免干滤纸吸收溶液中的溶剂,使结晶析出而堵塞滤纸孔),将溶液沿玻棒倒入,过滤时,漏斗上可盖上表面皿(凹面向下)减少溶剂的挥发,盛溶液的器皿一般用锥形瓶(只有水溶液才可收集在烧杯中)。
4、抽滤
抽滤前先熟悉布氏漏斗的构造及连接方式,将剪好的滤纸放入,滤纸的直径切不可大于漏斗底边缘,否则滤纸会折过,滤液会从折边处流过造成损失,将滤纸润湿后,可先倒入部分滤液(不要将溶液一次倒入)启动水循环泵,通过缓冲瓶(安全瓶)上二通活塞调节真空度,开始真空度可低些,这样不致将滤纸抽破,待滤饼已结一层后,再将余下溶液倒入,此时真空度可逐渐升高些,直至抽“干”为止。
停泵时,要先打开放空阀(二通活塞),再停泵,可避免倒吸。
5、结晶的洗涤和干燥
用溶剂冲洗结晶再抽滤,除去附着的母液。抽滤和洗涤后的结晶,表面上吸附有少量溶剂,因此尚需用适当的方法进行干燥。固体的干燥方法很多,可根据重结晶所用的溶剂及结晶的性质来选择,常用的方法有以卞几种:空气晾干的;烘干(红外灯或烘彩);用滤纸吸干;置于干燥器中干燥。
四、思考题
1、重结晶法一般包括哪几个步骤?各步骤的主要目的如何?
答:一般包括:(1)选择适宜溶剂,制成热的饱和溶液。(2)热过滤,除去不溶性杂质(包括脱色)。
(3)抽滤、冷却结晶,除去母液。(4)洗涤干燥,除去附着母液和溶剂。
2、重结晶时,溶剂的用量为什么不能过量太多,也不能过少?正确的应该如何?
答:过量太多,不能形成热饱和溶液,冷却时析不出结晶或结晶太少。过少,有部分待结晶的物质热溶时未溶解,热过滤时和不溶性杂质一起留在滤纸上,造成损失。考虑到热过滤时,有部分溶剂被蒸发损失掉,使部分晶体析出留在波纸上或漏斗颈中造成结晶损失,所以适宜用量是制成热的饱和溶液后,再多加20%左右。
3、用活性炭脱色为什单要待固体物质完全溶解后才加入?为什么不能在溶液沸腾时加入?
答:活性炭可吸附有色杂质、树脂状物质以及均匀分散的物质。因为有色杂质虽可溶于沸腾的溶剂中,但当冷却析出结晶体时,部分杂质又会被结晶吸附,使得产物带色。所以用活性炭脱色要待固体物质完全溶解后才加入,并煮沸5-10min。要注意活性炭不能加入已沸腾的溶液中,以一免溶液暴沸而从容器中冲出。
4、使用有机溶剂重结晶时,哪些操作容易着火?怎样才能避免呢?
答:有机溶剂往往不是易燃就是有一定的毒性,也有两者兼有的,操作时要熄灭邻近的一切明火,最好在通风橱内操作。常用三角烧瓶或圆底烧瓶作容器,因为它们瓶口较窄,溶剂不易发,又便于摇动,促使固体物质溶解。若使用的溶剂是低沸点易燃的,严禁在石棉网上直接加热,必须装上回流冷凝管,并根据其沸点的高低,选用热浴,若固体物质在溶剂中溶解速度较慢,需要较长时问,也要装上回流冷凝管,以免溶剂损失。
5、用水重结晶乙酰苯胺,在溶解过程中有无油状物出现?这是什么?
答:在溶解过程中会出现油状物,此油状物不是杂质。乙酰苯胺的熔点为114℃,但当乙酰苯胺用水重结晶时,往往于83℃就熔化成液体,这时在水层有溶解的乙酰苯胺,在熔化的乙酰苯胺层中含有水,故油状物为未溶于水而已熔化的乙酰苯胺,所以应继续加入溶剂,直至完全溶解。
6 、使用布氏漏斗过滤时,如果滤纸大于漏斗瓷孔面时,有什么不好?
答:如果滤纸大于漏斗瓷孔面时,滤纸将会折边,那样滤液在抽滤时将会自滤纸边沿吸入瓶中,而造成晶体损失。所以不能大,只要盖住瓷孔即可。
7、停止抽滤前,如不先拔除橡皮管就关住水阀(泵)会有什么问题产生?
答:如不先拔除橡皮管就关水泵,会发生水倒吸入抽滤瓶内,若需要的是滤液问题就大了。
8、某一有机化合物进行重结晶,最适合的溶剂应该具有哪些性质?
答:(1)与被提纯的有机化合物不起化学反应。(2)因对被提纯的有机物应具有热溶,冷不溶性质。(3)杂质和被提纯物质,应是一个热溶,一个热不溶。(4)对要提纯的有机物能在其中形成较整齐的晶体。(5)溶剂的沸点,不宜太低(易损),也不宜太高(难除)。(6)价廉易得无毒。
9、将溶液进行热过滤时,为什么要尽可能减少溶剂的挥发?如何减少其挥发?
答:溶剂挥发多了,会有部分晶体热过滤时析出留在滤纸上和漏斗颈中,造成损失,若用有机溶剂,挥发多了,造成浪费,还污染环境。
为此,过滤时漏斗应盖上表面皿(凹面向下),可减少溶剂的挥发。盛溶液的容器,一般用锥形瓶(水溶液除外),也可减少溶剂的挥发。
10、在布氏漏斗中用溶剂洗涤固体时应该注意些什么?
答:用重结晶的同一溶剂进行洗涤,用量应尽量少,以减少溶解损失。如重结晶的溶剂的熔点较高,在用原溶剂至少洗涤一次后。可用低沸点的溶剂洗涤,使最后的结晶产物易于干燥,(要注意此溶剂必须能和第一种溶剂互溶而对晶体是不溶或微溶的)。
实验七 萃 取
一、 实验的目的及基本要求
应用萃取可以从固体或液体混合物中提取所需要的物质,也可以用来洗去混合物中少量杂质。通常称前者为“抽提”或“萃取”,后者为“洗涤”,所以萃取是有机实验用来提取或纯化有机化合物常用操作之一。
通过实验让学生
(1)了解萃取分离的基本原理,乳化及破乳化。
(2)熟练掌握分液漏斗的选择及各项操作。
二、基本原理
萃取是利用物质在两种不互溶(或微溶)溶剂中溶解度或分配比的不同来达到分离、提取或纯化目的一种操作。
例:将含有有机化合物的水溶液用有机溶剂萃取时,有机化合物就在两液相之间进行分配。在一定温度下,此有机化合物在有机相中和在水相中的浓度之比为一常数,即所谓“分配定律”。
三、操作要点和说明
在实验中用得最多的是水溶液中物质的萃取,最常使用的萃取器皿为分液漏斗。
1、在使用分液漏斗前必须仔细检查:玻璃塞和活塞是否紧密配套。然后在活塞孔两边轻轻地抹上一层凡士林,插上活塞旋转一下,再看是否漏水。
2、将漏斗放于固定在铁架上的铁圈中,关好活塞,将要萃取的水溶液和萃取剂(一般为溶液体积的1/3)依次从上口倒入漏斗中,塞紧塞子。
3、取下分液漏斗,用右手撑顶住漏斗顶塞并握漏斗,左手握住漏斗活塞处,大拇指压紧活塞,把漏斗放平,旋转振摇,振摇几次后,将漏斗的上口向下倾斜,下部的支管指向斜上方(朝无人处),左手仍握在活塞支管处,用拇指和食指旋开活塞放气(释放漏斗内的压力),如此重复几次,将漏斗放回铁圈中静置,待两层液体完全分开后,打开上面的玻璃塞,再将活塞缓缓旋开,下层液体自活塞放出,然后将上层液体从分液漏斗的上口倒出(切记!)将水溶液倒回分波漏斗,再用新的萃取剂萃取。如此重复3-5次。
值得注意的是:
(1)分液时一定要尽可能分离干净,有时在两相间可能出现一些絮状物,也应同时放去(下层)。
(2)要弄清哪一层是水溶液。若搞不清,可任取一层的少量液体,置于试管中,并滴少量自来水,若分为两层,说明该液体为有机相,若加水后不分层则是水溶液。
(3)在萃取时,可利用“盐析效应”,即在水溶液中加入一定量的电解质(如氯化钠),以降低有机物在水中的溶解度,提高萃取效果。水洗操作时,不加水而加饱和食盐水也是这个道理。
(4)在萃取时,特别是当溶液呈碱性时,常常会产生乳化现象。这样很难将它们完全分离,所以要进行破乳,可加些酸。
4、萃取溶剂的选择要根据被萃取物质在此溶剂中的溶解度而定,同时要易于和溶质分离开。所以最好用低沸点的溶剂。一般水溶性较小的物质可用石油醚萃取。水溶性大的物质可用苯或乙醚;水溶性极大的物质可用乙酸乙酯。
5、分液漏斗使用后,应用水冲洗干净,玻璃塞和活塞用薄纸包裹后塞回去。
四、思考题
1、影响萃取法的萃取效率因素有哪些?怎样才能选择好溶剂?
答:(1)用同一分量的溶剂,分多次用少量溶剂来萃取,其效率高于一次用全量溶剂来萃取。(2)在萃取时,若在水溶液中先加入一定量的电解质(如NaCI)利用所谓“盐析效应”以降低有机化合物在水溶液中的溶解度,常可提高萃取效果。萃取溶剂的选择要根据被萃取物质在此溶剂中的溶解度而定。同时要易于和溶质分离开。所以最好选用低沸点的溶剂。
一般水溶性较小的物质可用石油醚萃取;水溶性较大的物质可用本或乙醚萃取;水溶性极大的物质可用乙酸乙酯萃取。
2、使用分液漏斗的目的何在?使用分液漏斗时要注意哪些事项?
答:分液漏斗主要应用于:(1)分离两种分层而不起作用的液体。(2)从溶液中萃取某种成分。(3)用水、酸或碱洗涤某种产品。(4)用来滴加某种试剂(即代替滴液漏斗)。
使用分液漏斗时应注意:
(1)不能把活塞上附有凡士林的漏斗放在烘箱内烘干。(2)不能用手拿分液漏斗的下端。(3)不能用手拿分液漏斗的进行分离。(4)玻塞打开后才能开启活塞。(5)上层的液体不要从分液漏斗下口放出。
3、乙醚作为一种常用的萃取剂,其优缺点是什么?
答:一般水溶性较大的物质可用乙醚或苯来萃取。乙醚沸点低,易除去是其优点。但沸点低,易燃,是其缺点。
实验八 簿层色谱(TLC法)
一、实验目的和基本要求
薄层色谱又叫薄板层析,是色谱法中的一种,是快速分离和定性分析少量物质的一种很重要的实验技术,属固-液吸附色谱,它兼备了柱色谱和纸色谱的优点,一方面适用于少量样品(几到几微克,甚至0.01微克)的分离;另一方面在制作薄层板时,把吸附层加厚加大,因此,又可用来精制样品,此法特别适用于挥发性较小或较高温度易发生变化而不能用气相色谱分析的质。此外,薄层色谱法还可用来跟踪有机反应及进行柱色谱之前的一种“预试”。
通过实验让学生
(1)初步掌握簿层色谱法的实验技术。
(2)学会用薄层色谱法来跟踪有机反应。
二、基本原理
这是利用吸附剂(硅胶、氧化铝等)对不同组分吸附能力的差异从而达到分离目的的方法。
三、操作要点和说明
薄层色谱法的整个过程包括以下步骤:
1、薄层板的制备 制薄层板的主要原料是吸附剂和粘结剂
吸附剂:最常用于 TLC的吸附剂为硅胶 GF254; 硅胶HF254。
粘结剂:一般用所羧甲基纤维素钠(CMC),也有用淀粉的。CMC 为粉状固体,用时先加水,水浴上熬成糊状,配成1%水溶液。
制板:(1)小板的制备:将硅胶加 1% CMC,调成桨状(在平铺玻璃板上能晃动但不能流动),将其涂在载玻片上( 75 X 25),为使其坦平,可将载玻片用手端平晃动,致坦平为止,放在干净平坦的台面上,晾干之后放入110℃烘箱活化1小时即可使用(一次可做很多块)。
(2)大板的制备:略
2、点样 点样用的毛细管为内径< lmm的管口平整的毛细管,将样品溶于低沸点的溶剂(乙醚、丙酮、乙醇、四氢呋喃等)配成1%溶液。
点样前,可先用铅笔在小板上距一端5mm处轻轻划一横线,作为起始线,然后用毛细管吸取样品在起始线上小心点样,如需重复点样,则应待前次点样的溶剂挥发后方可重点。若在同一块板上点几个样,样品点间距离为5mm以上。
3、展开 展开剂的选择主要根据样品的极性、溶解度和吸附剂的活性等因素来考虑。薄层的展开在密闭的容器中进行。先将选择的展开剂放入色谱器中(小板可用广口瓶代替),使色谱器内空气饱和5-10min,再将点好试样的薄层板放入色谱器中进行展开,点样的位置必须在展开剂液面之上,当展开剂上升到薄层的前沿(离前端 5-10mm)或多组分已明显分开时,取出薄层板放平晾干,用铅笔划溶剂前沿的位置后,即可显色。
4、显色 如果化合物本身有颜色,就可直接观察它的斑点。如果本身无色,可先在紫外灯光下观察有无荧光斑点(有苯环的物质都有),用铅笔在薄层板上划出斑点的位置;对于在紫外灯光下不显色的,可放在含少量碘蒸气的容器中显色来检查色点(因为许多化合物都能和碘成黄棕色斑点),显色后,立即用铅笔标出斑点的位置。
5、用 TLC跟踪有机反应 在同一块板上点上原料样和反应混合物样(均需配成稀溶液),按上述方法进行展开和显色,记下原料样的斑点位置和反应混合物样中相应斑点的位置和大小。过一定时间后,再取反应混合物样液点样、展开、显色,如发现反应混合液样中相应于原料斑点的位置处,无斑点或斑点变小,则说明反应已经完成或接近完成,所以依此可跟踪有机化学反应。这在有机合成上很有意义。
四、思考题
1、在一定的操作条件下为什么可利用Rf值来鉴定化合物?
答:薄层色谱是近年来发展起来的一种微量、快速而简单的色谱法。因为色谱法的基本原理是利用混合物中各组分在某一物质中的吸附或溶解性能(即分配)的不同或其它亲和作用性能的差异,使混合物溶液流经该种物质,进行反复的吸附或分配等作用,从而将各组分分离。所以可利用Rf值来鉴定化合物。化合物不同,其Rf值是不同的。
2、在混合物薄层色谱中,如何判定各组分在薄层上的位置?
答:薄层色谱展开剂的选择和柱色谱一样,主要根据样品的极性,溶解度和吸附剂的活性等因素来考虑。凡溶剂的极性越大,则对化合物的洗脱力也越大,也就是说Rf值也越大,(如果样品在溶剂中有一定溶解度)。因为根据溶剂的极性,化合物的极性及Rf值,可知各组分在薄层板上的位置。
3、展开剂的高度若超过了点样线,对薄层色谱有何影响?
答:这样所点试样将被溶剂所洗脱,薄层色谱无法展开。
实验九 溴乙烷的制备
一、目的和要求
1、学习从结构上相对应的醇为原料制备一卤代烷的原理和方法。
2、学习低沸点物蒸馏的基本操作。
3、掌握萃取的原理和方法。
二、反应原理
 用乙醇、溴化纳及硫酸作用是制备溴乙烷常用的方法。
用乙醇、溴化纳及硫酸作用是制备溴乙烷常用的方法。
 NaBr+H2SO4 HBr+NaHSO4
NaBr+H2SO4 HBr+NaHSO4
C2H5OH+HBr C2H5Br+H2O
溴乙烷的沸点很低(38.4℃)
上式的制备反应是一个可逆反应,通常可以采用增加其中一种反应物的浓度或设法移走产物使溴乙烷及时离开反应体系的方法,使平衡向右移动。本实验正是两种措施并用,以使反应顺利完成。
若H2SO4浓度太大,又会引起一系列副反应。

 H2SO4+HBr SO2+H2O+Br2
H2SO4+HBr SO2+H2O+Br2
2C2H5OH C2H5-O-C2H5 +H2O
 C2H5OH CH2=CH2+H2O
C2H5OH CH2=CH2+H2O
反应混合物中水量太少。HBr气体容易在操作时逃逸而使反应不完全。但是由于这个反应是可逆的,水的用量太大,也不利于反应的完全,因此,反应混合物中水的量是需要讨论的第一个关键问题。
三、药品试剂,操作步骤(略)
四、操作要点及注意事项
1、反应开始时,加热不可太猛,否则蒸出的油层还有较深的黄色,这可能是HBr受硫酸氧化所致,再者加热太猛时,反应瓶中的物质起泡严重,有可能冲到冷凝管中。
2、溴乙烷沸点低(38.4℃),产物极易挥发损失,因此,反应完毕后,应将粗产物暂保留在接受器水层以下。先准备好后面所用的干燥仪器(小锥形瓶、分液漏斗)、蒸馏装置,然后进行分离,精制、洗涤……
3、粗产物中的水分一定要分离干净,然后在冷却下加入浓H2SO4,如果水分不尽,加浓H2SO4时将发热,使产物挥发、损失。另一方面浓H2SO4的作用是洗涤除去粗产物中所含的乙醇、乙醚等。
4、本实验是第一次使用分液漏斗,教师要突出的讲分液漏斗分液的原理,使用范围及操作注意事项。
五、思考题
1、溴乙烷的制备中浓H2SO4洗涤的目的何在?
答:除去副产物、乙醚、乙烯和原料乙醇
2、溴乙烷沸点低(38.4℃),实验中采取了哪些措施减少溴乙烷的损失?
答:①反应中加入少量的水,防止反应进行时发生大量的泡沫,减少副产物乙醚的生成和避免HBr 的挥发。②C2H5Br在水中的溶解度小(1:100)常在接受器预放冷水并将接液管的末端浸入水中。 ③分离时尽可能将水分离干净,否则用浓H2SO4洗涤时会产生热量,导致产物的挥发。④蒸馏的速度要慢,否则蒸气来不及冷凝而损失。 ⑤选择高效冷凝,各接头不漏气
实验十 1-溴丁烷的制备
一、目的和要求
1、学习以溴化钠、浓硫酸和正丁醇为原料制备1-溴丁烷的原理和方法。
2、练习带有吸收有害气体装置的回流加热操作。
二、反应原理
 NaBr + H2SO4 HBr + NaHSO4
NaBr + H2SO4 HBr + NaHSO4
 n-C4H9OH + HBr n-C4H9Br + H2O
n-C4H9OH + HBr n-C4H9Br + H2O
上述反应是一个可逆反应,本实验采用增加HBr的量来增大正丁醇的转化率。若反应体系温度过高可能发生下列一系列副反应:

因此,反应体系温度的控制是本实验的关键。
三、药品试剂,操作步骤( 略)
四、操作重点和注意事项
1、在圆底烧瓶中加入10ml水,然后加入14ml浓硫酸,等浓硫酸冷至室温后才加入溴化钠。
2、正溴丁烷必须蒸完,否则会影响产率,这可以从以下几个方面判断:
(1)溜出液是否由浑浊变为澄清。
(2)反应瓶上层油层是否消失。
(3)取一试管收集几滴溜出液,加水振摇,观察有无油珠出现。
3、产物处理过程中,加入浓硫酸是为了除去未反应的正丁醇,否则它与正溴丁烷形成共沸物而难以除去。
五、思考题
1、反应后的粗产物中含有哪些杂质?各步洗涤的目的何在?
答:杂质有正丁醚、未反应的正丁醇和氢溴酸,加入浓硫酸是为了除去未反应的正丁醇,有机相依次用水、饱和碳酸氢钠和水洗涤的目的是除去未反应的硫酸、氢溴酸和过量的碳酸氢钠。
2、用分液漏斗洗涤产物时,正溴丁烷时而在上层,时而在下层,若不知道产物的密度,可用什么简便的方法加以判断?
答:在分液漏斗中加入一些水,体积增大的那一层是水层,另一层是有机层。
实验十一 环己烯的制备
一、目的和要求
1、学习以浓磷酸催化环已醇脱水制环已烯的原理和方法。
2、初步掌握分馏和水浴蒸馏的基本操作。
二、反应原理
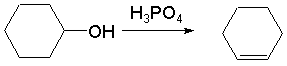
实验室小量制备常采用醇,酸催化脱水的方法。整个反应是可逆的。为了促使反应完成,必须不断的将反应生成沸点低的烯烃蒸出来。
由于高浓度的酸会导致烯烃的聚合,分子间的失水及碳化,故常伴有副产物的生成。
三、药品试剂,操作步骤( 略)
四、操作重点及注意事项
1、本反应是可逆的,故采用了在反应过程中将产物的反应体系分离出来的办法,推动反应向正反应方向移动,提高产物的产率。
2、为了使产物以共沸物的形成分出反应体系 ,又不夹带原料环已醇。本实验采用分馏装置。
3、条件的控制及注意事项
⑴环己醇粘度大,尤其低温,量筒内的环已醇很难倒净,会影响产率。
⑵磷酸有一定的氧化性,加磷酸要摇勺后加热,否则会被氧化。
⑶加热一段时间后,再逐渐蒸出产物,调节滴加速度,保持反应速度大于蒸出速度,才能使馏继续进行,控制柱顶的温度稳定在71℃不波动。
⑷反应终点的判断可参考下面几个参数:a)反应进行40分钟左右;b)分馏出的环 已烯-水煮沸物达到理论值;c)反应瓶中出现白雾;d)柱顶温度下降后又升到85℃以上。
五、思考题
1、 采用分馏装置制备环己烯,要控制分馏柱顶端温度不超过90℃,为什么?
答:主要由反应产物(b.p.82.98℃)和几种恒沸物和生成水的沸点决定的。
2、在粗制的环 已醇加入精盐,使水层饱和的目的是什么?
答:进行盐析,提高产率
实验十二 正丁醚的制备
一、目的和要求
1、掌握醇分子间脱水制醚的反应原理和实验方法。
2、学习使用分水器的实验操作。
二、反应原理
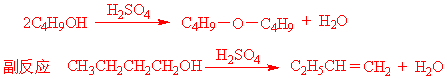
三、药品试剂,操作步骤( 略)
四、操作重点及注意事项
1、醇转变成醚如果是定量进行的话,那么反应中生成的水可以定量的算出,本实验就是根据理论计算出生成水的体积,然后由装满水的分水器中给予扣除,当反应生成的水、正好充满分水器,而将从冷凝后的醇正好溢流反回反应瓶中,从而达到自动分离指示反应完全的目的。
2、制正丁醚的温度要严格控在135℃以下,否则易产生大量的副产物正丁烯。
3、在碱洗时不要剧烈振动,否则易生成乳浊液难于分离。
五、思考题
本实验用50%H2SO4洗涤粗产品,为什么不要浓H2SO4洗涤粗产品?
答:用50%H2SO4洗粗产品主要洗除副产物丁烯和未反应完的原料正丁醇。若用浓H2SO4洗涤则连同正丁醚都一起洗去了。
实验十三 苯乙酮的制备
一、实验目的及要求
学习Friedel-Cragto酰化法,制备芳香酮的原理和方法。
二、反应原理
付-克酰基化,是制备芳酮的主要方法。在无水三氯化铝的存在下,酰氯、酸酐与活泼的芳基化合物反应得到高产率的芳酮。
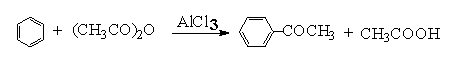
三、药品试剂、操作步骤( 略)
四、操作重点及注意事项
1、滴加苯乙酮和乙酐混合物的时间以10min为宜,滴的太快温度不易控制。
2、无水三氯化铝的质量是本实验成败的关键,以白色粉末打开盖冒大量的烟,无结块现象为好。
3、苯以分析纯为佳,最好用纳丝干燥24小时以上再用。
4、粗产物中的少量水,在蒸馏时与苯以共沸物形式蒸出,其共沸点为69.4℃,这是液体化合物的干燥方法之一.
五、思考题
1、在苯乙酮的制备中,水和潮气对本实验有何影响?在仪器装置和操作中应注意哪些事项。
答:破坏试剂影响产率,导致失败因三个试剂都属无水。应采取措施:⑴药品仪器均需干燥。⑵回流冷凝管上装一个干燥管。⑶整个装置密合不漏气。
2、反应完成后,为什么要加入浓盐酸和冰水的混合液?
答:加酸的目的是使苯乙酮析出,其反应如下:

实验十四 乙酰苯胺的制备
一、目的和要求
1、掌握苯胺乙酰化反应的原理和实验操作;
2、掌握固体有机化合物提纯的方法--重结晶。
二、反应原理
胺的酰化在有机合成中有着重要的作用。作为一种保护措施,一级和二级芳胺在合成中通常被转化为它们的乙酰基衍生物以降低胺对氧化降解的敏感性,使其不被反应试剂破坏;同时氨基酰化后降低了氨基在亲电取代反应(特别是卤化)中的活化能力,使其由很强的第Ⅰ类定位基变为中等强度的第Ⅰ类定位基,使反应由多元取代变为有用的一元取代,由于乙酰基的空间位阻,往往选择性的生成对位取代物。

三、药品试剂、操作步骤( 略)
四、操作重点及注意事项
制备完成后要趁热将瓶内混合物倒入盛有冷水的烧杯中,否则固体析出沾在瓶壁上不易处理。
五、思考题
1、 本反应为什么要控制分馏柱顶端温度在105℃?
答:主要由原料CH3COOH(b.p.118℃)生成物水(b.p.100℃)的沸点所决定 。控制在105℃这样可以保证原料CH3COOH充分反应而不被蒸出,生成的水立即移走促使反应向生成物方向移动,有利于提高产率。
2、用苯胺作原料进行苯环上的一些取代时,为什么常常要先进行酰化呢?
答:防止苯胺的氧化,减少副产物生成
实验十五 三苯甲醇的制备
一、目的和要求
1、了解格氏试剂的制备、应用和进行格氏反应的条件。
2、掌握搅拌、回流、萃取蒸馏(包括低沸点物蒸馏)等操作。
二、反应原理
三苯甲醇可通过格氏试剂反应用下列任一方法制取
方法一、二苯甲酮与苯基溴化镁反应

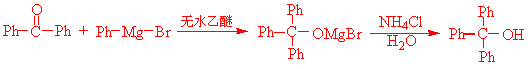
方法二、苯甲酸乙酯与苯基溴化镁的反应
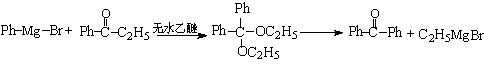
得到苯甲酮后按方法一进行。
三、药品试剂、操作步骤( 略)
四、操作重点及注意事项
1、所用仪器、药品必须经过严格的干燥处理,否则反应很难进行,并可使生成的格氏试剂分解。
2、卤代烃与镁的作用很难发生通常温热或用一小粒碘作催化剂,以促使反应开始。
3、滴加的速度太快,反应过于激烈,不易控制,并会增加副产物的生成。
4、为了使反应易于发生,故搅拌应在反应开始以后进行,若5min后反应仍不开始,可用温水浴或直接加入一小粒碘促使反应开始。
五、思考题
1、本实验有哪些可能的副反应?如何避免?
答:本实验的副反应如下:
 RMgX + H2O RH + Mg(OH)X
RMgX + H2O RH + Mg(OH)X
 2RMgX + O2 2ROMgX
2RMgX + O2 2ROMgX
 RMgX + RX R-R + MgX2
RMgX + RX R-R + MgX2
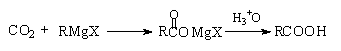
增加干燥防护装置。
2、本实验中溴苯加的太快或一次加入有什么不好?
答:实验是一个放热反应,一次加入或滴加速度太快,易造成反应过于激烈,不易控制并会增加副产 物的生成
实验十六 环己酮的制备
一、实验目的和要求
1、学习铬酸氧化法制环己酮的原理和方法。
2、通过第二醇转变为酮的实验 ,进一步了解醇和酮之间的联系和区别。
二、反应原理
实验室制备脂肪或脂环醛酮,最常用的方法是将伯醇和仲醇用铬酸氧化。铬酸是重要的铬酸盐和40-50%硫酸的混合物。
仲醇用铬酸氧化是制备酮的最常用的方法。酮对氧化剂比较稳定,不易进一步氧化。铬酸氧化醇是一个放热反应,必须严格控制反应的温度,以免反应过于激烈。

三、药品试剂,操作步骤( 略)
四、操作重点及注意事项
1、本实验是一个放热反应,必须严格控制温度。
2、本实验使用大量乙醚作溶剂和萃取剂,故在操作时应特别小心,以免出现意外。
3、环己酮在31℃水解度为2.4g /100ml水中。加入粗盐的目的是为了降低溶解度,有利于分层。
五、思考题
用铬酸氧化法环己酮的制备实验,为什么要严格控制反应温在55~60℃之间,温度过高或过低有什么不好?
答:本反应是一个放热反应,温度高反应过于激烈,不易控制,易冲出,温度过低反应不易进行,导致反应不完全
17 8-羟基喹啉的制备
一、实验目的和要求
1.学习合成8-羟基喹啉的原理和方法
2.巩固回流加热和水蒸汽蒸馏等基本操作
二、反应原理
Skraup反应,是合成杂环化合物喹啉及其衍生物最重要的方法,它是用苯胺与无水甘油,浓硫酸及弱氧化剂硝基化合物等一起加热而得,为了避免反应过于剧烈,常加入FeSO4作为氧的载体。浓硫酸的作用使甘油脱水成丙烯醛,并使苯胺与丙烯醛的加成物脱水成环。硝基化合物则将1.2-二氢喹啉氧化成喹啉,本身被还原成芳胺也可以参加缩合。反应中所用的硝基化合物,要与芳胺的结构相对应,否则会导致产生混合物。8-羟基喹啉形成的过程如下:

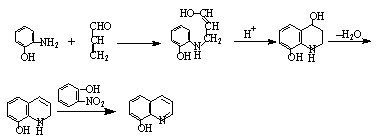
三、药品试剂、操作步骤( 略)
四、操作重点及注意事项
1、所用甘油含水量不超过0.5%(d=1.26)如果甘油含水量较大时,则喹啉的产量不高,可将其加热到18℃,冷却在100℃左右放入盛有浓H2SO4的干燥器中备用。
2、此反应系放热反应,要严格控制反应温度以免冲出容器。
3、8-羟基喹啉既溶于碱又溶于酸而成盐,且成盐后不被水蒸汽蒸馏为此必须小心中和,严格控制在pH=7-8之间,当中和恰当时,瓶内析出的8-羟基喹啉沉淀最多。
4、粗产物用4:1(体积比)C2H5OH:H2O,25ml混合液重结晶时,由于8-羟基喹啉难溶于冷水,于放置滤液中,慢慢滴入无离子水,即有8-羟基喹啉不断析出结晶。
五、思考题
1、为什么第一次水蒸汽蒸馏要在酸性条件进行第二次水蒸汽蒸馏要在中性条件下进行?
答:在酸性条件下使其成盐,不被蒸出这样可以除去未作用的硝基苯酚,第二次水蒸汽蒸馏使其在中性条件下,被水蒸汽带出。
 2、在反应中如用对甲基苯胺作原料应得到什么产物?硝基化合物应如何选择?
2、在反应中如用对甲基苯胺作原料应得到什么产物?硝基化合物应如何选择?
 产物为 硝化物
产物为 硝化物
18 甲基橙的制备
一、实验目的和要求
1、通过甲基橙的制备掌握重氮化反应和偶合反应的操作。
2、巩固盐析和重结晶的原理和操作。
二、反应原理
甲基橙是指示剂,它是由对氨基苯磺重氮盐与-N,N一二甲基苯胺的醋酸盐,在弱酸介质中偶合得到的,偶合先得到的是红色的酸性甲基橙,称为酸性黄,在碱性中酸性黄转变为甲基橙的钠盐,即甲基橙

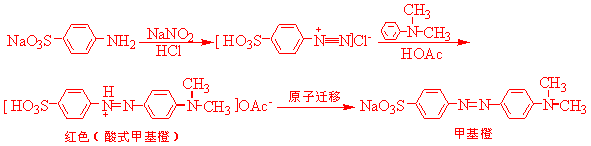
三、药品试剂 、操作步骤( 略)
四、操作重点及注意事项
1、对氨基苯磺酸是两性化合物酸性比碱性强以酸性内盐形成存在。但重 氮时,又要在酸性溶液中进行,因此生氨时,首先将对氨基苯磺酸与碱作用变成水溶性较大的细盐。
2、重氮化过程中,严格控制温度很重要,反应温度高于5℃则生成的重 氮盐易水解用的酚,降低产率,导致失败。
3、粗产品呈碱性,温度稍高,易使产物变质,颜色变深,温的甲基橙受日光照射,亦会颜色变淡,通常在55-78℃烘干。
五、思考题
1、本实验中重氮盐的制备为什么要控制在0-5℃中进行?偶合反应为什么要在弱酸介质中进行?
答:温度高了易产生重 氮盐分解,使偶联反应不能发生。
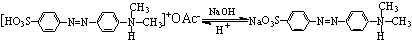 2、甲基橙在酸碱中介质中变色的原因,并用反应式表示之。
2、甲基橙在酸碱中介质中变色的原因,并用反应式表示之。
答:
乙酰乙酸乙酯的制备
一、实验目的和要求
1、了解乙酰乙酸乙酯的制备原理和方法。
2、掌握无水操作及减压蒸馏等操作。
二、反应原理
含α-活泼氢的酯在碱性催化剂存在下,能与另一分子的酯发生酯缩合反应,生成β-羰基酸酯,乙酰乙酸乙酯,就是通过这一反应来制备的,当 用金属钠作偶合剂时,真正的催化剂是钠与乙酸乙酯残留的少量乙醇作用产生的醇钠,一旦反应开始,乙醇就可以不断生成并和金属钠继续作用。如果使用高纯度的乙酸乙酯和金属钠反而不能发生缩合反应。反应经历了以下平衡:
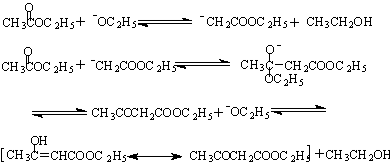
三、药品试剂、操作步骤( 略)
四、操作重点及注意事项
1、本实验的关键:是所用仪器必须是干燥的,所用的乙酸乙酯必须是无水的。水的存在金属钠易与水反应生成放出氢气及大量的热易导致燃烧和爆炸。NaOH的存在易使乙酸乙酯水解成乙酸钠,更重要的是水的存在使金属钠消耗难以形成碳负离子中间体,导致实验失败。
2、金属钠遇水易燃烧爆炸,在空气中易氧化,故在称量压丝和切成片的过程中,操作要迅速。金属钠的颗粒大小,直接影响酯缩合反应的速度,最好将压细机出口的钠丝,直接装入盛有乙酸乙酯的烧瓶内。
3、缩合反应是一个放热反应,用小火加热控制上升蒸气在 回流冷凝管的第一个小球下,以免挥发损失。反应完毕后得一透明带有绿色荧光的桔红色液体,有时被检出少量黄白色固体(乙酰乙酸乙酯的烯醇或钠盐)
4、由于金属纳遇水易爆炸,燃烧,不宜用水溶加热。
5、乙酰乙酸乙酯,在常压蒸馏时很易分解,其分解产物为“失水乙酸”这样会影响产率,故采用减压蒸馏法。
6、本实验的产率通常根据金属钠的用量来计算的要注意本实验,从头至尾尽可能在1-2天内完成,如果间隔时间太长,会因失水乙酸的生成,而降低产量。
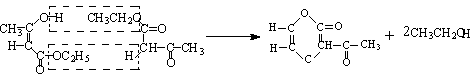
五、思考题
1、乙酰乙酸乙酯在合成上有什么用途?烷基取代乙酰乙酸乙酯与稀碱和浓碱作用将分别得到什么产物?
答:活泼亚甲基可以被其它基团取代,如另一方面乙酰乙酸乙酯在不同反应条件下,可以分解成酸和酮受稀碱作用按酮式分解, 浓碱作用按酸或分解。
2、如何利用乙酰乙酸乙酯合成下列化合物。
⑴2-康酮 ⑵△-P基-2-巳酮 ⑶2.6-2庚酮
答:略。