附件1
医疗器械临床评价技术指导原则
(征求意见稿)
一、编制目的
医疗器械临床评价是指注册申请人通过临床文献资料、临床经验数据、临床试验等信息对产品是否满足使用要求或者适用范围进行确认的过程。本指导原则旨在为注册申请人进行临床评价提供技术指导,同时也为食品药品监督管理部门对临床评价资料的审评提供技术参考。
二、法规依据
(一)《医疗器械监督管理条例》(国务院令第650号);
(二)《医疗器械注册管理办法》(国家食品药品监督管理总局令第4号);
(三)医疗器械临床试验质量管理相关规定。
三、适用范围
本指导原则适用于第二类、第三类医疗器械注册申报时的临床评价工作,不适用于按医疗器械管理的体外诊断试剂的临床评价工作。
四、列入《免于进行临床试验的医疗器械目录》产品的临床评价要求
1
对于列入《免于进行临床试验的医疗器械目录》(以下简
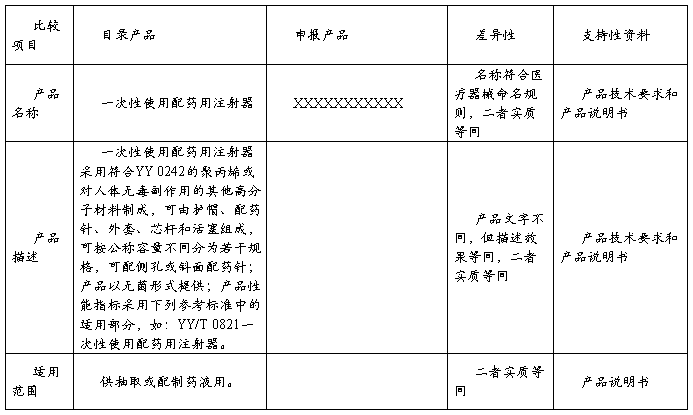
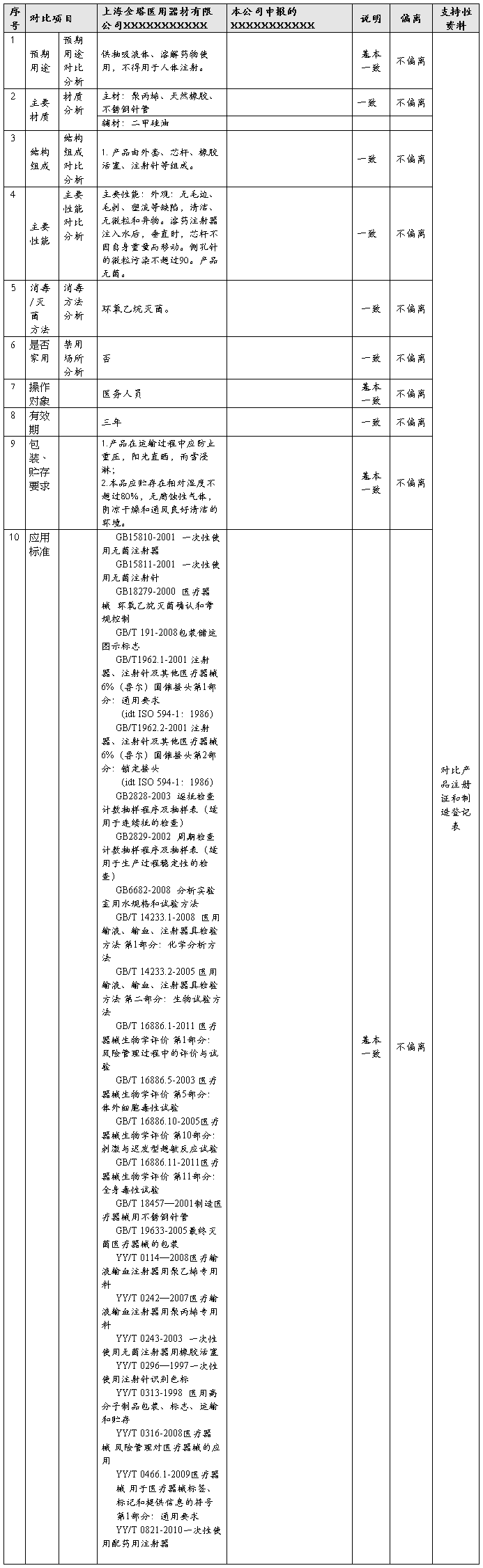
称《目录》)产品的临床评价,注册申请人需将申报产品与《目录》所述内容进行对比以判定申报产品是否为列入《目录》产品。
列入《目录》产品是指与《目录》所述的产品名称、产
品描述、预期用途具有等同性的产品。注册申请人对申报产品的相关信息与《目录》所述内容进行对比,论述其相同性和差异性。当二者的差异性对产品的安全有效性不产生影响时,认为二者具有等同性。
注册申请时需提交的临床评价资料为申报产品与《目录》产品的对比表及附件(格式见附件1)。
五、通过同品种医疗器械临床数据进行临床评价的一般
要求
(一) 基本原则
通过同品种医疗器械临床数据进行临床评价应全面、客
观,需将收集的临床性能和安全性数据、有利的和不利的数据均纳入分析。临床评价的深度和广度、需要的数据类型和数据量应与产品的设计、关键技术、预期用途和风险程度相适应,也应与非临床研究的水平和程度相适应。同品种医疗器械临床数据的证据强度不应低于进行临床试验获得的数据。
…… …… 余下全文